Pulmonary arterial hypertension
Author information
1Univ. Paris-Sud, Faculté de Médecine, Kremlin-Bicêtre F-94270, France
2AP-HP, DHU TORINO, Centre de Référence de l’Hypertension Pulmonaire Sévère, Service de Pneumologie et Réanimation Respiratoire, Hôpital Bicêtre, Le Kremlin-Bicêtre F-94270, France
3INSERM U999, Labex LERMIT, Hypertension Artérielle Pulmonaire: Physiopathologie et Innovation Thérapeutique, Centre Chirurgical Marie Lannelongue, Le Plessis-Robinson F-92350, France
4Pulmonary Hypertension Service, Royal Brompton Hospital, London SW3 6NP, UK
Corresponding author.
#Contributed equally.David Montani: rf.phpa.tcb@inatnom.divad; Sven Günther: rf.phpa.tcb@rehtnug.nevs; Peter Dorfmüller:
rf.dusp-u@rellumfrod.retep; Frédéric Perros: moc.liamg@sorrep.cirederf; Barbara Girerd: rf.phpa.tcb@drerig.arabrab; Gilles Garcia: rf.phpa.tcb@aicrag.sellig; Xavier Jaïs: rf.phpa.tcb@siaj.reivax; Laurent Savale: rf.phpa.tcb@elavas.tnerual; Elise Artaud-Macari: rf.phpa.tcb@iracam-duatra.esile; Laura C Price: moc.liamtoh@ecirpcarual; Marc Humbert: rf.phpa.tcb@trebmuh.cram; Gérald Simonneau: rf.phpa.tcb@uaennomis.dlareg; Olivier Sitbon: rf.phpa.tcb@nobtis.reivilo
Abstract
Pulmonary arterial hypertension (PAH) is a chronic and progressive disease leading to right heart failure and ultimately death if untreated. The first classification of PH was proposed in 1973. In 2008, the fourth World Symposium on PH held in Dana Point (California, USA) revised previous classifications. Currently, PH is devided into five subgroups.
Group 1 includes patients suffering from idiopathic or familial PAH with or without germline mutations. Patients with a diagnosis of PAH should systematically been screened regarding to underlying mutations of BMPR2 gene (bone morphogenetic protein receptor type 2) or more rarely of ACVRL1 (activine receptor-like kinase type 1), ENG (endogline) or Smad8 genes. Pulmonary veno occusive disease and pulmonary capillary hemagiomatosis are individualized and designated as clinical group 1′.
Group 2 ‘Pulmonary hypertension due to left heart diseases’ is divided into three sub-groups: systolic dysfonction, diastolic dysfonction and valvular dysfonction.
Group 3 ‘Pulmonary hypertension due to respiratory diseases’ includes a heterogenous subgroup of respiratory diseases like PH due to pulmonary fibrosis, COPD, lung emphysema or interstitial lung disease for exemple.
Group 4 includes chronic thromboembolic pulmonary hypertension without any distinction of proximal or distal forms.
Group 5 regroup PH patients with unclear multifactorial mechanisms. Invasive hemodynamic assessment with right heart catheterization is requested to confirm the definite diagnosis of PH showing a resting mean pulmonary artery pressure (mPAP) of ≥ 25 mmHg and a normal pulmonary capillary wedge pressure (PCWP) of ≤ 15 mmHg.
Definition and classification
Pulmonary arterial hypertension (PAH) is defined by right-heart catheterization (RHC) showing precapillary pulmonary hypertension with a mean pulmonary artery pressure (mPAP) of >25 mmHg and a normal pulmonary artery wedge pressure (PCWP) of <15 mmHg [1,2]. The classification of pulmonary hypertension (PH) has gone through a series of changes since the first classification proposed in 1973 which designated only two categories, primary pulmonary hypertension or secondary PH, depending on the presence or absence of identifiable causes or risk factors [3,4]. In 1998, a second World Symposium on PH was held in Evian (France) and this classification attempted to create categories of PH that shared similar pathogenesis, clinical features and therapeutic options [5]. This classification allowed defining homogenous groups of patients to conduct clinical trials and to obtain approval for specific PAH therapies worldwide. In 2003, the third World Symposium on PH (Venice, Italy) did not propose major changes. However, the terms idiopathic PAH, familial PAH, and associated PAH were introduced. The other prominent change was to move pulmonary veno-occlusive disease (PVOD) and pulmonary capillary hemangiomatosis (PCH) from separate categories into a single subcategory of PAH.
In 2008, the fourth World Symposium on PH held in Dana Point (California, USA) and the consensus of an international group of experts was to revise previous classifications in order to accurately reflect published data, as well as to clarify some areas that were unclear. In 2013, the fifth World Symposium on PH held in Nice (France) and proposed only minor modifications, however, since the definite conclusions of this symposium were not yet published, we presented the Dana Point classification of PH (Table 1).
Table 1
Table 1
Diagnostic classification of pulmonary hypertension
| 1. Pulmonary arterial hypertension (PAH) | 1.1 Idiopathic |
| 1.2 Heritable | |
| 1.3 Drugs and toxins induced | |
| 1.4 Associated with (APAH): | |
| 1.4.1 Connective tissue disease | |
| 1.4.2 Infection with human immunodeficiency virus | |
| 1.4.3 Portal hypertension | |
| 1.4.4 Congenital heart disease | |
| 1.4.5 Schistosomiasis | |
| 1.4.6 Chronic haemolytic anaemia | |
| 1.5 Persistent pulmonary hypertension of the newborn | |
| 2. Pulmonary hypertension with left heart disease | 2.1 Systolic dysfunction |
| 2.2 Diastolic dysfunction | |
| 2.3 Valvular disease | |
| 3. Pulmonary hypertension due to lung diseases and/or hypoxia | 3.1 Chronic obstructive pulmonary disease |
| 3.2 Interstitial lung disease | |
| 3.3 Other pulmonary diseases with mixed restrictive and obstructive pattern | |
| 3.4 Sleep-disordered breathing | |
| 3.5 Alveolar hypoventilation disorders | |
| 3.6 Chronic exposure to high altitude | |
| 3.7 Developmental abnormalities | |
| 4. Chronic thromboembolic pulmonary hypertension | |
| 5. PH with unclear and/or multifactorial mechanisms | 5.1 Haematological disorders: myeloproliferative disorders, splenectomy. |
| 5.2 Systemic disorders: sarcoidosis, pulmonary Langerhans cell histiocytosis, lymphangioleiomyomatosis, neurofibromatosis, vasculitis | |
| 5.3 Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders | |
| 5.4 Others: tumoral obstruction, fibrosing mediastinitis, chronic renal failure on dialysis |
Epidemiology
Information relative to the natural history of PAH was derived from a national registry conducted in the USA in the early 1980s, which included 187 patients with idiopathic PAH followed for up to 5 years. This study characterized the disease and confirmed its poor prognosis with a median survival of 2.8 years [105,106]. Better understanding of pathophysiological mechanisms of PAH has led to the development of novel therapeutic strategies in the last decade which improved quality of life, exercise capacity and survival of these PH patients [107].
In France, a national multicenter registry prospectively collected data from 674 adults with PAH from October 2002 to October 2003 and followed these patients during a 3-year period [108]. This prospective study has shown more up to date data on the epidemiology of PAH. This registry has confirmed the female predominance in most subtypes of PAH with a female/male sex ratio (SR) of 1.9 for all PAH patients, and particularly in idiopathic PAH (SR 1.6), familial PAH (SR 2.2) and anorexigen-associated PAH (SR 14.9) [108]. The mean age of PAH patients in this cohort was 50 years with a quarter of patients older than 60 years underlining the possibility of developing PAH in all ages. In this assessed cohort, 39.2% of patients had idiopathic PAH and 3.9% familial PAH. In the subgroup of PAH, where PAH was associated with other conditions, 15.3% had connective tissue disease, 11.3% congenital heart diseases, 10.4% portal hypertension, 9.5% anorexigen-related PAH and 6.2% HIV infection [108]. In France, the low estimation of the prevalence of PAH and idiopathic PAH are respectively of 15 cases and 5.9 cases/million adult inhabitants. The low estimation of PAH incidence is 2.4 cases/million adult inhabitants/year. This study conducted five years after the withdrawal of fenfluramine derivatives underlines that anorexigen-associated PAH remains an important medical problem nowadays.
Recent prospective data in consecutive patients with idiopathic, familial or anorexigen-associated PAH patients followed-up for a period of three years (56 incident and 298 prevalent cases) showed better survival rates than historical cohort. For incident cases, estimated survival rates were 85.7%, 69.6% and 54.5% at one, two and three year of follow-up, respectively. In combined mixed population (incident patients and prevalent patients diagnosed within three years before study entry), estimated one, two and three year survival was 82.9%, 67.1% and 58.2%, respectively. Parameters significantly associated to improved survival were female gender, preserved 6-MWD and normal cardiac output on RHC [109].
Data from the REVEAL registry (US Registry to Evaluate Early and Long-Term PAH Disease Management) are similar concerning the one year survival rate estimated to 91%. Variables independently associated with increased mortality included pulmonary vascular resistance >32 Wood units, NYHA functional class IV, men older than 60 years and familiy history of PAH [110].
Genetics
It is now well known that PAH can be either sporadic or clustered in families [111-114]. In 1954, Dresdale published a detailed description of a family that included three related subjects with severe PAH of unknown aetiology. Because of physician’s awareness of the familial occurrence of the disease, other cases of familial PAH were described. The National Institute of Health Registry (NIH Registry) provided the first estimate of this condition, and evaluated that at least 6% of individuals diagnosed with sporadic PAH have a family history of the disorder. In the French PAH registry, 26 cases of familial PAH were identified in the cohort of 674 patients, which corresponds to a prevalence of 3.9% of all cases of PAH and a proportion of 7.3% of familial PAH in the subgroup of idiopathic, familial or anorexigen-associated PAH [108].
Studies of genealogies of familial PAH advanced our understanding that the disease segregates an autosomal dominant trait with a markedly reduced penetrance, since only 10-20% of necessary carriers of the mutation will develop PAH [115,116]. In 2000, linkage analysis in PAH affected families found mutations within the bone morphogenetic protein receptor type 2 gene (BMPR2) [117,118]. BMPR2 gene encodes for a type 2 receptor member of the transforming growth factor beta (TGF-β) family. Nowadays, germline BMPR2 mutations are detected in 58-74% of PAH patients with a family history of the disease, and in 3.5-40% of so called idiopathic PAH patients [6,11,108,119-123]. The observation of the development of PAH in patients displaying hereditary hemorrhagic telangiectasia (HHT), an autosomal dominant vascular dysplasia, allowed us to identify two other PAH predisposing genes: ACVRL1 (Activin A receptor type II-like kinase 1) and ENG (endoglin) genes. Mutations in these two genes are infrequent in PAH but are frequently identified in HHT [120,124-130].
Moreover, recent reports described two PAH patients carriers of a mutation in Smad8 gene, one PAH patient carrier of a Smad1 mutation and one PAH patient carrier of a Smad5 mutation [8,9]. BMPR2 gene, ACVRL1, ENG, Smad8, Smad1 and Smad5 genes are encoding proteins which are involved in the TGF-β signaling pathway. More recently, using whole exome sequencing in a large PAH family, Austin et al. demonstrated the involved of mutations in caveolin-1 gene in the development of PAH [10] They confirmed their findinds identifying a caveolin-1 mutation in an unrelated PAH patient. Caveolin-1 protein is necessary for the formation of calveola, which are crucial for joining membrane receptors and initiating cellular signaling cascade such as TGF-β signalling pathway. This supports the hypothesis that mutations in genes involved in the TGF-β signaling pathway may be a trigger for pulmonary vascular remodeling. Moreover, this signaling pathway controls growth, differentiation and apoptosis of various cell types like pulmonary vascular endothelial cells (ECs) and smooth muscle cells (SMCs). Thereby, mutations in genes involved in the TGF-β signaling pathway may be responsible for abnormal proliferation of pulmonary vascular SMCs and may promote ECs apoptosis, which might lead to the selection of apoptosis resistant cells and formation of plexiform lesions, the hallmark of idiopathic PAH [131,132].
Pathophysiology
PAH is a disease which affects small pulmonary arteries. It is characterized by vascular obstruction leading to progressive increase in vascular resistance. This increases right ventricular afterload and consequently results in right ventricular failure. Intima and media proliferation and its consequent pulmonary vascular obstruction are considered to be the key element in the pathogenesis of PAH. Vasoconstriction, vascular remodeling and thrombosis are factors that increase pulmonary vascular resistance in PAH [107,137]. These processes involve a multitude of cellular and molecular elements (Figure 1).
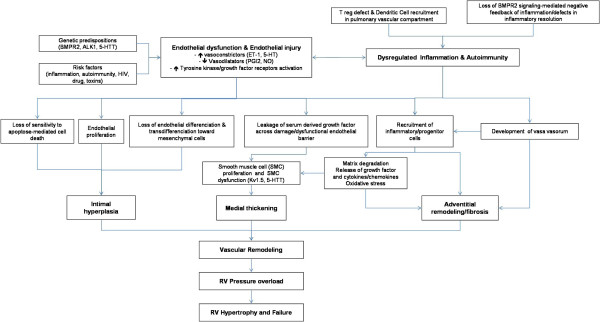
Pathophysiology of PAH. The pulmonary vascular remodeling responsible for PAH is the consequence of closely intertwined predisposing and acquired factors. Thoses pathological elements affect all three layers of precapillary pulmonary arteries leading to intimal hyperplasia, medial thickening and adventitial remodeling/fibrosis. Intra- but also extra-pulmonary cells, such as inflammatory and progenitor cells, are suspected to play a role in this remodeling. This increases right ventricular afterload and consequently results in right ventricular failure.
Cellular factors
Proliferation of smooth muscular cells in the small peripheral pulmonary arteries is a common characteristic in all forms of PAH. In hypoxic models, fibroblasts of the adventitia migrate to the media and intima, where proliferation and production of matrix proteins are observed [138]. Neovascularization, mainly of the adventitia, occurs concomitantly to the thickening of the vascular walls [139].
In response to certain stimuli, endothelial cells abnormally proliferate to form plexiform lesions in several forms of PAH. Plexiform lesions consist of endothelial cells, matrix proteins and fibroblasts and obliterate the vascular lumen [140]. The stimuli for endothelial proliferation is still unknown but several factors have been incriminated such as hypoxia, inflammation, shear stress, drugs, viral infections and genetic susceptibility. Extrapulmonary cells may also participate in the vascular remodeling responsible for PAH. Indeed, fibrocytes and c-kit + cells are mobilized from the bone marrow, and may differentiate into vascular cells and/or produce pro-angiogenic factors to participate in the pathogenesis of PAH [141,142]. The CXCL12/CXCR4 axis may play an important role in the pulmonary recruitment of these circulating progenitors and can be therapeutically targeted [143].
Molecular factors
Many authors consider pulmonary vasoconstriction as an early event in the process of PAH. Vasoconstriction has been associated with an abnormal function or expression of potassium channels and with endothelial dysfunction [107]. Endothelial dysfunction results in a decreased production of vasodilators such as nitric oxide (NO) and prostacyclin and an increased production of vasoconstrictors such as endothelin-1 [153].
Prostacyclin (prostaglandin I2) is a potent pulmonary vasodilator that acts via the cyclic adenosine monophosphate (cAMP) pathway. It inhibits the proliferation of smooth muscle cells and decreases platelet aggregation. Production of prostacyclin is reduced in endothelial cells of patients with PAH [154]. PAH therapy based on prostacyclin and its derivates have proven efficacy both hemodynamically and in clinical trials. NO is also a pulmonary vasodilator which acts via the cyclic guanosine monophosphate (cGMP) pathway. To increase pulmonary vasodilatation dependant on NO, a recent therapeutic strategy has targeted type 5 phosphodiesterase which degrades cGMP. Sildenafil or tadalafil, type 5 phosphodiesterase inhibitors, have proven their efficacy in patients with PAH [155]. Vasoactive intestinal peptide (VIP) is a neurotransmitter that has systemic and pulmonary vasodilator properties. It also inhibits smooth cell proliferation and decreases platelet aggregation and acts via the activation of the cAMP and cGMP systems [156]. Low plasmatic concentrations of VIP have been measured in pulmonary arteries of patients with PAH.
Histopathology: vascular changes
Vascular remodelling in pulmonary arterial and venous hypertension typically involves the small pulmonary vessels. Muscular arteries of less than 500 μm display a thickening within the intimal, medial and adventitial compartment. When involved, pulmonary veins of the interlobular septa and smallest preseptal venules show fibrous obliteration and / or muscularization. Even the smallest vascular level may be involved: patients with pulmonary venous hypertension frequently present focal proliferation of alveolar capillaries. The different histological pattern is presented here below.
Arterial lesions
Isolated medial hypertrophy
This abnormality of the vessel wall can be observed in all subgroups of PAH and may even be encountered in other forms of PH, e.g. in mitral valve stenosis. The lesion corresponds to a smooth muscle cell proliferation and / or recruitment within the tunica media; the histological criterion of hypertrophy / hyperplasia is fulfilled, when the diameter of a single medial layer, delimited by its internal and external elastic lamina, exceeds 10 per cent of the arteries cross-sectional diameter (Figure 2A). Isolated hypertrophy of the medial layer may be considered as an early and even reversible event as it has been shown in PH due to hypoxia in high altitude [171]. However, medial hypertrophy is usually associated with other PAH-lesions.
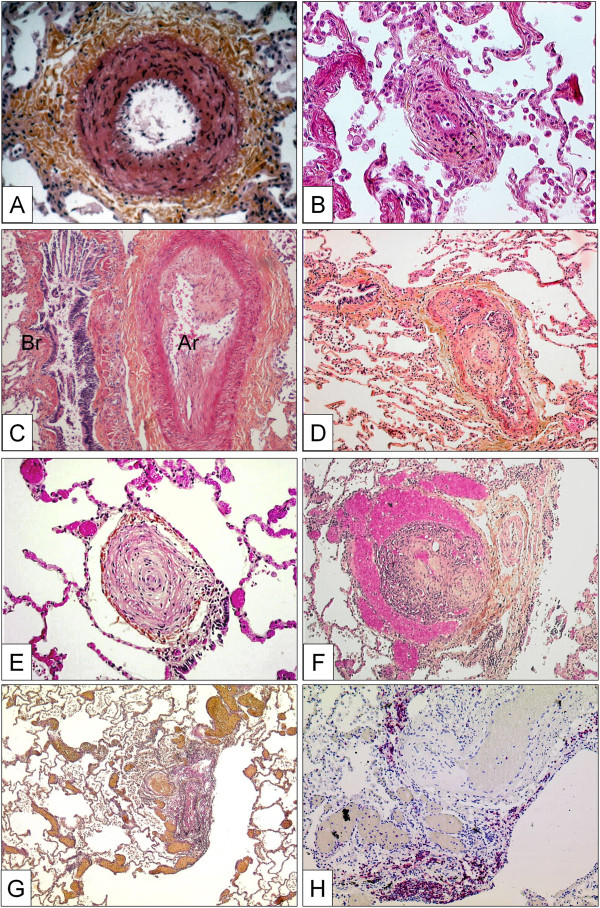
Pulmonary arteries of the muscular type displaying obstructive arteriopathy in lungs of patients with PAH. A Medial hypertrophy with smooth muscle cell proliferation and pronounced adventitial fibrosis. Magnification x200, Weigert-hematoxylin-phloxine-saffron staining (WHPS). B Concentric non-laminar intimal fibrosis comprising numerous myofibroblasts (arrows). C Eccentric intimal fibrosis corresponding to organized thrombotic material. Br: bronchus, Ar: pulmonary artery. Magnification x100, HES staining. D Thrombotic lesion, so called “colander-like lesion”, with partial recanalization by microvessels. Note the similarity to plexiform lesions (F). Magnification x100, HES. E Concentric laminar intimal fibrosis, so called „onion-skin lesion“. Magnification × 200, HES. F Plexiform lesion with proliferation of small sinusoid-like vessels on a fibrotic matrix. Note surrounding dilated vessels. Magnification x100, HES. G Multiple dilation lesions being the sentinel of the centrally located plexiform lesion. Magnification × 40, Elastica-van-Gieson staining (EvG). H The same plexiform lesion after immunohistochemical staining with anti-CD3, a T-lymphocytic marker. Note the perivascular distribution of the inflammatory infiltrate. Magnification x100.
Concentric and eccentric non-laminar intimal fibrosis
Fibrotic lesions of the intimal layer are frequent in PAH-diseased lungs. The intima may be thickened by proliferation and recruitment of fibroblasts, myofibroblasts and other connective tissue cells, and consequently by the interstitial deposition of collagen (Figure 2B, C). In a purely descriptive approach, this thickening may be uniform and concentric, or focally predominating and eccentric. However, the eccentric intimal thickening is frequently observed in cases with thrombotic events and could represent residues of wall-adherent, organized thrombi. Thrombotic lesions, or so called in situ thrombosis, are a frequent pattern in different PAH-subgroups: organization and recanalization of totally occluding thrombotic material may lead to bizarre, fibrotic multi-channel lesions (so called “colander-like” lesions) which can be easily confounded with proliferative complex lesions (see below) (Figure 2D). Frequently, adventitial fibrosis is associated to intimal modifications (Figure 2A, B).
Venous and venular lesions (Pulmonary veno-occlusive disease and pulmonary hemangiomatosis)
A clear-cut differentiation between pre-and post-capillary pulmonary vascular lesions is sometimes difficult to make: lesions frequently concern veins and arteries in lungs of patients with PVOD, and vice-versa veins may be strongly involved in some subgroups of PAH. This is not contradictory because the clinical approach to ‘difficult-to-treat’ PAH-groups may be similar to clinical management of PVOD and PVOD-patients are frequently treated – under great precaution – with pulmonary arterial dilators, e.g. prostanoids. Recent reports, for example, indicate that CTD associated PAH, classically being considered as pre-capillary PH, simultaneously displays a PVOD-like pattern in histology [178,179]. Like in PVOD, the observed post-capillary lesions concern septal veins and pre-septal venules and usually consist of a loose and pauci-cellular and cushion-like intimal fibrosis that may totally occlude the lumen. A muscularization of both, septal veins and pre-septal venules may be observed (Figure 3A, B). Importantly, occult pulmonary hemorrhage regularly occurs in patients displaying PVOD. This particularity, which is certainly due to the post-capillary bloc, is of diagnostic importance, as bronchio-alveolar lavage can reveal an occult hemorrhage. The degree of hemorrhage can be evaluated semi-quantitatively and qualitatively using the Golde Score, which takes number and Perls-Prussian-Blue staining-degree of intra-alveolar siderophages into consideration (Figure 3C) [180]. Pulmonary capillary hemangiomatosis has been historically described as an aggressive capillary proliferation with patchy distribution within the pulmonary parenchyma: alveolar septa are thickened by 3 to 4 capillary layers, and infiltration of venous and bronchiolar structures with secondary occlusion may be present (Figure 3D).
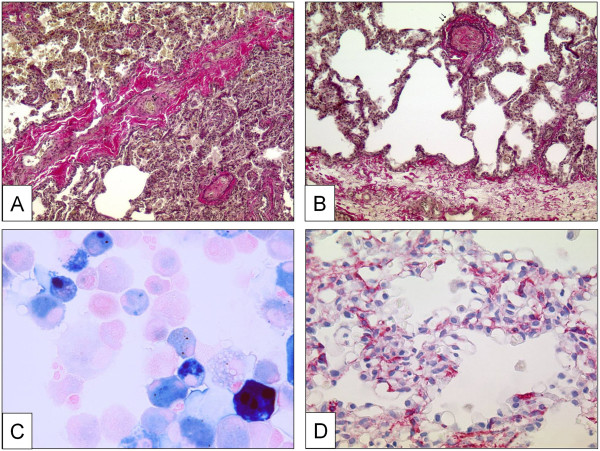
Pulmonary veins with obstructive venopathy in lungs of patients with PVOD and a case of pulmonary capillary hemangiomatosis. A Longitudinally dissected septal vein with asymmetric intimal and partially occlusive fibrosis. Note the intra-alveolar hemorrhage due to the post-capillary block on the upper half of the photograph. Magnification × 100, EvG. B Pre-septal venule with occlusive intimal fibrosis. Magnification × 100, EvG. C Bronchio-alveolar lavage in a PVOD-patient. Perls-Prussian-Blue staining. Note the siderophages displaying gradually different color-shades (see text). Magnification × 400. D Excessively proliferating alveolar capillaries in a patient with pulmonary capillary hemangiomatosis. Note protrusion of ectatic lumina into the alveoli. Magnification × 200, anti-CD31 staining.
Clinical description
Symptoms and clinical signs of PH
There is no pathognomonic clinical sign of PH. Clinical presentation is related either to right heart failure or to associated diseases. Persistant dyspnea on exertion is the most frequent symptom; and it is present in almost patients even in the presence of mild hemodynamic abnormalities [1,182]. Dyspnea usually starts insiduously and is often neglected by patients which explain the delay of around two years in establishing the diagnosis of PH. The New York Heart Association (NYHA) provides a classification system for the clinicial evaluation of dyspnoea. Four categories are proposed to classify patients in functional classes (FC) based on how much they are limited during physical activity; the limitations/symptoms are in regard to normal breathing (Table 3).
Table 3
Modified New York Heart Association (NYHA) classification for pulmonary hypertension
| CLASS I | Patients with pulmonary hypertension but without resulting limitation of physical activity. Ordinary physical activity does not cause undue dyspnoea or fatigue, chest pain, or near syncope. |
| CLASS II | Patients with pulmonary hypertension resulting in slight limitation of physical activity. They are comfortable at rest. Ordinary physical activity causes undue dyspnoea or fatigue, chest pain, or near syncope. |
| CLASS III | Patients with pulmonary hypertension resulting in marked limitation of physical activity. They are comfortable at rest. Less than ordinary activity causes undue dyspnoea or fatigue, chest pain, or near syncope. |
| CLASS IV | Patients with pulmonary hypertension with inability to carry out any physical activity without symptoms. These patients manifest signs of right heart failure. Dyspnoea and/or fatigue may even be present at rest. Discomfort is increased by any physical activity. |
However, at time of diagnosis, 70% of patients are in NYHA FC III or IV. Chest pain, light-headedness and syncope may occur, particularly during physical efforts and are major signs of disease severity. Palpitations are frequent during physical efforts and may reveal true cardiac arrhythmias. Other symptoms of PAH include fatigue and weakness. Hemoptysis may complicate PAH and could be life-threatening, justifying embolization of dilated bronchial arteries. Hoarseness of the voice may occasionally be noted and is due to compression of the left laryngeal nerve by the dilated pulmonary artery (Ortner’s syndrome).
Diagnostic methods
The diagnostic process of PAH requires a series of investigations that are intended to make the diagnosis, to clarify the clinical class of PH and the underlying type of PAH and to evaluate the functional and hemodynamic impairment [183]. The detection of PH requires investigations including electrocardiogram, chest radiograph and trans-thoracic echocardiogram. Other conditions which can induce PH will be identified by tests such as pulmonary function tests, arterial blood gases, ventilation and perfusion lung scan, high resolution computed tomography (HR-CT) of the chest and pulmonary angiography. Additional investigations are required for evaluation of PAH severity including exercise testing and hemodynamics. Additional imaging may clarify underlying lung abnormalities. Finally, right heart catheterisation confirms the definite diagnosis.
Management of PAH
General measures
Physical activities
Peripheral vasodilatation or increased cardiac demand will put PAH patient at risk of acute cardiac failure and syncope. Thus, we traditionally advise against extreme physical activity. Patients are taught to stay active while adapting effort according to their symptoms. Nevertheless, the appropriate level of physical activity is difficult to define. To date, only few studies have evaluated to effect of cardio-respiratory rehabilitation in PAH. One of this program involved three weeks of inpatient rehabilitation followed by three month training at home with phone call supervision. No modification on cardiac hemodynamic was observed on echocardiography but 6-MWD and quality of life were improved. As proposed in ERS/ESC guidelines, more data are required before appropriate recommendations can be made [15].
Altitude and hypoxia
As hypoxic vasoconstriction may be an aggravating factor in PAH, stays at altitude above 1500-2000 meters without supplemented oxygen and air flight in unpressurized cabin should be avoided [15]. Chronic hypoxia (PaO2 <60 mmHg) warrants oxygen therapy for symptoms and to avoid PAH deterioration.
Pregnancy and contraception
The hemodynamic and hormonal modifications occurring during pregnancy and peripartum period can lead to severe, and sometimes fatal, right heart failure [197,198]. Pregnancy is considered to be associated with high rate of mortality (30-50%) in PAH patients [15]. Thus, pregnancy is contraindicated in women affected by PAH. Consequently, contraception is strongly recommended in PAH women of childbearing age [197,198]. Combined estrogen-progestin oral contraceptive is theoretically contraindicated because their pro-thrombotic activity. Therefore, mechanical contraception (intra-uterine device for example) or surgical sterilization should be proposed. Nethertheless, pregnancy should be managed with specific PAH therapies and planned elective delivery in expert centres [15].
Anaesthesia and surgery
Hypotension induced by anaesthetic drugs, and hemodynamic insults following surgical and anaesthetic interventions are generally poorly tolerated by PAH patients, with a high procedural morbidity and mortality. Consequently, it is recommended in general that PAH patients avoid unnecessary procedures. When required, these should be highly planned with the multidisciplinary team, either within, or in close liaison with, the expert centers, in order to coordinate surgical and medical interventions [199].
Proscribed drugs
Vasoconstrictors used in cold medication to relieve nasal congestion should be avoided in PAH patients. Beta-blockers have been shown to be deleterious to PAH patients because they prevent the important adaptive physiologic response that allows preservation of adequate cardiac output. To discontinue such drugs in a patient with PAH may lead, by itself, to important clinical and hemodynamic improvement [200].
Non specific drugs
Diuretics
Diuretics are one of the most important treatments in the setting of PAH because right heart failure leads to fluid retention, hepatic congestion, ascites and peripheral edema. Right ventricular overload is part of clinical symptoms and has been associated with a poor prognosis in PAH [201]. Diuretics and salt-free diet relieve hypervolemia and associated symptoms. Whether this strategy improves prognosis is unknown. Dose adjustment of diuretics is needed, based on clinical and hemodynamic findings. Renal function and blood chemistry should be monitored to avoid renal failure or dyskalemia [192].
Oral anticoagulation
Pathology specimens from PAH patients may show in situ thrombosis and thrombi recanalisation. Only few studies support anticoagulation treatment in PAH (mostly retrospective or not randomized) [202,203]. Current recommendations propose oral anticoagulation aiming for targeting an International Normalized Ratio (INR) between 1.5 and 2.5. Although the somewhat sparse evidence base is derived exclusively from idiopathic, heritable and PAH due to anorexigens, anticoagulation has been generalised to all patient groups, given the absence of contraindication. Anticoagulation is usually not recommended in porto-pulmonary hypertension because of the risk of esophageal variceal haemorrhage. In patients with systemic sclerosis, oral anticoagulation can be difficult to manage because of their high risk of bleeding, especially from the gastrointestinal tract. Variceal ligation is a preventive option in these cases. Long-term oral anticoagulation is essential in CTEPH with an INR which is recommended to be between 2 and 3.
Digitalis
Digoxin has been suggested as part of PAH therapy in the past because it produces an acute increase in cardiac output [204], although its efficacy is unknown in PAH. Therefore it is usually proposed in PAH associated with atrial tachyarrhythmias [192].
Calcium channel blockers
Calcium channel blockers (CCB) are indicated in patients with a positive vasodilatation challenge test after inhaled NO. CCB are vasodilators and were initially introduced in the 1980’s as part of PAH therapy to counteract vasoconstriction that has traditionally been assumed to be a preponderant mechanism in PAH. In the 1990’s, Rich and colleagues showed in an open prospective study that high dose of calcium channel blockers (nifedipine 90 to 240 mg/day or diltiazem 360 to 900 mg/day) significantly improve prognosis in patients with an acute vasodilation response [203]. Sitbon et al. showed that only 12.6% out of the patients could be treated by CCB according to criteria’s establish by Rich and co-workers [203]. Moreover, only half of them maintained actual long-term benefit defined as NYHA FC I or II at one year without the need for additional treatment [205]. In this study, a decrease in mean PAP > 10 mmHg to reach a value < 40 mmHg, with a stable or increased cardiac output, during acute vasodilator test was predictive of long-term response to CCB. Contrarily, CCB must be avoided in the absence of acute vasoreactivity because of the risk of significantly reduced cardiac output and systemic blood pressure [205]. The choice of CCB is based upon the patient’s heart rate; nifedipine and amlodipine were preferred in the presence of relative bradycardia and diltiazem in the presence of tachycardia. Doses of CCB used in this setting are relatively high, 120-240 mg de nifedipine, 240-270 mg for diltiazem, and up to 20 mg for amlodipine [15]. If the patient does not show a correct response (NYHA FC I or II with marked hemodynamic improvement), additional PAH specific therapy should be added. Of note, CCB is contraindicated in PVOD because of the high-risk of life threatening pulmonary edema, even in the presence of acute vasodilator response that can be observed in the same proportion as in idiopathic PAH [53].
PAH-specific therapies
Better understanding in pathophysiological mechanisms of PH over the past quarter of a century has led to the development of medical therapeutics, even though no cure for pulmonary arterial hypertension exists. Several specific therapeutic agents were developed for the medical management of PAH including prostanoids, endothelin receptor antagonists and phosphodiesterase type 5 inhibitors. Furthermore, emerging treatments such as tyrosine kinase inhibitors, soluble guanylate cyclase activators (riociguat) and prostacyclin receptor agonists (selexipag) are currently being evaluated in PAH.
Prostanoids
Endothelium-derived prostaglandin I2 (PGI2), or prostacyclin, is an arachidonic acid produced by endothelial cells. Prostacyclin is a powerful systemic and pulmonary vasodilator and an inhibitor of platelet aggregation through the increase in intracellular cyclic adenosine monophosphate (cAMP) [206,207]. Moreover, prostacyclin plays an important role in antiproliferative, antithrombotic, antimitogenic and immunomodulatory activity [208]. Prostanoids are a family of prostacyclin analogues available in intravenous, subcutaneous, or inhaled form.
Epoprostenol In the 1980s, intravenous epoprostenol was the first prostanoid evaluated in PAH [209]. As the half-life of epoprostenol is <5 minutes, it requires an indwelling central venous catheter which is connected to an infusion pump for continuous intravenous administration. Treatment with epoprostenol is complex, uncomfortable and expensive and cannot be considered as an ideal treatment despite its evident clinical benefit. Common side effects associated with treatment include headache, flushing, jaw pain and gastrointestinal disturbance [201,210]. The efficacy of continuous i.v. administration of epoprostenol has been tested in three unblinded RCTs in idiopathic PAH [207,211] and PAH associated with systemic sclerosis [212].
Barst and colleagues [183] showed improvement in exercise capacity with an increase in 6-MWD of 47meters after 12 months of epoprostenol treatment in 81 patients with idiopathic PAH. Moreover, a significant improvement of survival was observed (no death at three months of treatment in the group with epoprostenol against 8 deaths in the group receiving conventional treatment including diuretics and anticoagulants [213]. Long-term persistence of efficacy has also been shown [201,214] in idiopathic PAH, as well as in other associated PAH [215,216] and in non operable CTEPH [217]. Long term effectiveness has never been evaluated prospectively but retrospective analysis comparing patients treated by intravenous epoprostenol with data from patients treated with conventional therapy find meaningful clinical benefit for patients in NYHA FC III or IV [201,214,218,219]. Functional class, hemodynamic parameters and long term survival were all improved in the group treated with i.v. epoprostenol.
Epoprostenol had initially been proposed as a bridging therapy for lung transplantation but it is currently regarded as the treatment of choice for patients in NYHA FC IV. If treatment with epoprostenol is necessary, it will be started at low dose of 2-4 ng/kg/min and is gradually increased to 10-16 ng/kg/min according to side effects [201]. Treatment interruption secondary to pump dysfunction or the rupture of the catheter, although rare, can induce serious adverse events [201]. Because of its route of administration, central venous catheter bloodstream infections can occur, and should be systematic searched in the context of unexplained clinical deterioration.
Treprostinil Due to complications related to the central venous catheter used for the i.v. administration of epoprostenol, other prostacyclin agonists have been developed. Treprostinil is a prostacyclin analogue which benefits from a longer half-life of 58–83 minutes by subcutaneous administration [220]. It is delivered by a minipump similar to those used for insulin [221]. A multicenter randomized trial evaluated subcutaneous administrated treprostinil versus placebo over three months in patients in NYHA functional class II-IV suffering from idiopathic PAH, PAH related to CHD with a shunt or PAH associated with CTD [221]. Patients treated with treprostinil increased 6-MWD and had benefits like quality of life, pulmonary hemodynamics and improvement of clinical symptoms. Unfortunately, local side-effects such as pain and inflammation at the injection site are present in the majority of patients treated with treprostinil which often lead to limitation of increasing dose or treatment cessation. Intravenous treprostinil is only licensed for the use in the USA and provides the advantage of less frequent need for drug reservoir replacement [220]. Inhaled treprostinil was examined in a placebo-controlled TRIUMPH-1 study [222], but results of this formulation were not convincing and this product is not yet licensed outside the USA.
Inhaled Iloprost Iloprost is a prostacyclin analogue administered by inhalation or the intravenous route. The pulmonary vasodilating effects of inhaled iloprost lasts nearly 45 minutes, therefore six to nine inhalations daily are needed, with each of them requiring approximately 30 minutes [223,224]. Common side effects of treatment were cough, flushing, jaw pain and headache [223]. In the AIR study [223] conducted in at 37 European pulmonary hypertension centers, study participants (idiopathic PAH, PAH associated to systemic sclerosis, anorexigen associated PAH or non operable CTEPH) in functional class III or IV were assessed during a three month period. Patients received a daily inhalation of 2.5 μg or 5.0 μg of iloprost 6 or 9 times a day or placebo. After 3 months of treatment, 17% of patients receiving iloprost reached the combined endpoint of improvement in functional class and 10% gain in 6-MWD as compared to 5% in the placebo group.
Endothelin receptors antagonists
Endothelin-1 (ET-1) is a potent vasoconstrictor and therefore plays an important role in the pathogenesis of PAH. In addition, it is responsible for smooth muscle cell proliferation [158]. Elevated ET-1 plasma levels are found in patients suffering from PAH and are correlated with poor prognosis. There are two existing isoforms of ET-1 receptors: endothelin A (ETRA) and endothelin B (ETRB). Activation of ETRA and ETRB on pulmonary artery smooth muscle cells induce proliferation and vasoconstriction, whereas activation of ETRB on pulmonary endothelial cells leads to release of NO and prostacyclin and participate to the clearance of circulating ET-1.
Bosentan Bosentan is an oral active dual ETRA and ETRB antagonist. Bosentan has been evaluated in PAH (idiopathic, associated with CTD, and Eisenmenger’s syndrome) in five RCT’s (Pilot, BREATHE-1, BREATHE-2, BREATHE-5, and EARLY) that have shown improvement in exercise capacity, functional class, haemodynamic, echocardiographic and Doppler variables, and time to clinical worsening [107,225–228]. The first placebo-controlled study included 32 patients affected by idiopathic PAH or scleroderma presenting with PAH showing significant exercise improvement with a gain of 76 meters after three months of treatment with bosentan as compared to placebo [225].
The BREATHE 1 study confirmed the efficacity of treatment with Bosentan in more than 200 assessed patients in NYHA functional class III or IV compared to placebo in a three month trial. After three months of treatment with bosentan, improvements in NYHA functional class were observed in 42% of patients receiving bosentan compared with 30% in the placebo arm. 6-MWD was improved overall to 44 meters in favor of bosentan. Furthermore, delayed time to clinical worsening was also noted as well as better results in dynpnea scores [226]. One accepted common side effect of treatment is increase in liver enzymes; this is why monthly monitoring of liver transaminases is mandatory in all patients receiving bosentan. Treatment is started at a dose of 62.5 mg twice a day and increased to the dose of 125 mg twice daily after one month of treatment.
Subsequently published data of treatment with bosentan suggested persistent improvements in pulmonary hemodynamics, exercise capacity and modified NYHA functional class and possibly survival rate of patients [229–231]. Results from the EARLY study (double-blind, randomised controlled 6 months trial) showed that the effect of the dual endothelin receptor antagonist bosentan in patients with mildly symptomatic PAH could be beneficial for PAH patients in NYHA functional class II [228].
Ambrisentan Ambrisentan is a selective ETA receptor antagonist administrated once daily at a dose of 5 mg or 10 mg. Two large RCTs (ARIES I and II, i.e. Ambrisentan in Pulmonary Hypertension, Randomized, Double blind, Placebo-controlled Multicenter, Efficacy Studies I and II) have demonstrated efficacy on symptoms, haemodynamics and time to clinical worsening of patients with idiopathic PAH and associated to CTD and HIV infection [232]. An extension study of the ARIES study is the recently published ARIES-E study by Klinger and colleagues [233]. They followed patients for a mean period of 60 weeks in where patients underwent hemodynamic evaluation. The authors concluded that treatment with ambrisentan leads to hemodynamic stability in PAH patients.
Phosphodiesterase type-5 inhibitors
NO is a potent pulmonary arteries SMC relaxant that disposed vasodilator activity through up-regulation of its associated down-stream signalling molecule, cyclic GMP (cGMP), metabolism of which is dependent on the activation of a number of PDEs [208]. Phosphodiesterase type 3, 4 and 5 are the three main types of this enzyme found in pulmonary artery contractive cells. PDE-5 is the most abundantly expressed isoform in pulmonary circulation which was confirmed by several experimental investigations showing a beneficial effect of PDE-5 inhibitors on vascular remodelling and vasodilatation [234,235].
Sildenafil Sildenafil is an oral PDE-5 inhibitor that is available in Europe since 2005 for PAH patients in functional class II-III whereas this drug is licensed in Canada and the USA for patients in functional class II-IV. Basis for the authorization of this drug in the setting of PAH was a large randomized, placebo-controlled trial in which different doses of sildenafil were assessed in 278 patients presenting with idiopathic PAH, PAH related to CTD or congenital systemic to pulmonary shunts surgically corrected. The majority of study patients were in functional class II-III. After three months of treatment, the mean placebo-adjusted changes in 6-MWD for 20 mg, 40 mg and 80 mg doses of sildenafil were 45 meters, 46 meters and 50 meters, respectively. Furthermore, significant hemodynamic and functional class improvements were noted in every sildenafil group as compared to placebo. Common side effects of treatment with sildenafil include headache, flushing and dyspepsia but no hepatic enzymes increase was noted as compared with endothelin receptor antagonists. Long term extension data from 222 patients who completed one year of sildenafil monotherapy with a dose of 80 mg three times daily showed encouraging results with a gain in 6-MWD, suggesting a durable treatment effect [236]. However, sildenafil approval in Europe is currently limited to 20 mg three times a day. No data is currently available on the long-term efficacy of this lower dosage.
Tadalafil Another PDE-5 inhibitor is tadalafil which was granted for use in patients with PAH in Europe and North America in 2009. Galiè and colleagues [237] assessed in the PHIRST trial 405 randomly assigned patients who were either treatment naïve or already receiving bosentan therapy to placebo or one of the several proposed doses of tadalafil 2.5 mg or 10 mg or 20 mg or 40 mg once daily for a period of three months. At study completion, patients receiving tadalafil showed an overall mean placebo-correlated increase in 6-MWD of 33 meters [208]. Thus, this increase was dose-dependent, with only the 40 mg dose achieving the prespecified value for statistical significance for improvement. Data analysis of comparative hemodynamic data from 93 patients who underwent repeat RHC has significant reduction in mPAP and PVR under treatment with tadalafil. Barst and co-authors [238] underlined favourable effects with tadalafil 40 mg among patients receiving background bosentan, although improvements were less marked compared with treatment naïve cohort.
Authors’ contributions
DM, SG, PD, FP, BG, GG, XJ, LS, EAM, LCP, MH, GS, OS participated in drafting the manuscript. All authors read and approved the final manuscript.
References
References
- Rubin LJ. Primary pulmonary hypertension. N Engl J Med. 1997;336(2):111–7. [PubMed] [Google Scholar]
- Simonneau G. et al.Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S43–54. [PubMed] [Google Scholar]
- Wagenvoort CA, Wagenvoort N. Primary pulmonary hypertension: a pathological study of the lung vessels in 156 clinically diagnosed cases. Circulation. 1970;42:1163–84. [Google Scholar]
- Hatano S, Strasser T. Primary Pulmonary Hypertension. Report on a WHO meeting. October 15–17, 1973. Geneva: WHO; 1975. [Google Scholar]
- Fishman AP. Clinical classification of pulmonary hypertension. Clin Chest Med. 2001;22(3):385–91. vii. [PubMed] [Google Scholar]
- Cogan JD. et al.High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174(5):590–8. [PMC free article] [PubMed] [Google Scholar]
- Aldred MA. et al.BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum Mutat. 2006;27(2):212–3. [PubMed] [Google Scholar]
- Shintani M. et al.A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J Med Genet. 2009;46(5):331–7. [PubMed] [Google Scholar]
- Nasim MT. et al.Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat. 2011;2011:2011. [PubMed] [Google Scholar]
- Austin ED. et al.Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet. 2012;5(3):336–43. [PMC free article] [PubMed] [Google Scholar]
- Machado RD. et al.Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat. 2006;27(2):121–32. [PubMed] [Google Scholar]
- Thomson JR. et al.Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-ß family. J Med Genet. 2000;37:741–5. [PMC free article] [PubMed] [Google Scholar]
- Chaouat A. et al.Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax. 2004;59(5):446–8. [PMC free article] [PubMed] [Google Scholar]
- Trembath RC. et al.Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2001;345(5):325–34. [PubMed] [Google Scholar]
- Galie N. et al.Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34(6):1219–63. [PubMed] [Google Scholar]
- Simonneau G. et al.Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):5S–12S. [PubMed] [Google Scholar]
- Souza R. et al.Pulmonary arterial hypertension associated with fenfluramine exposure: report of 109 cases. Eur Respir J. 2008;31(2):343–8. [PubMed] [Google Scholar]
- Rich S. et al.Anorexigens and pulmonary hypertension in the United States: results from the surveillance of North American pulmonary hypertension. Chest. 2000;117(3):870–4. [PubMed] [Google Scholar]
- Walker AM. et al.Temporal trends and drug exposures in pulmonary hypertension: an American experience. Am Heart J. 2006;152(3):521–6. [PubMed] [Google Scholar]
- Frachon I. et al.Benfluorex and unexplained valvular heart disease: a case–control study. PLoS One. 2010;5(4):e10128. [PMC free article] [PubMed] [Google Scholar]
- Savale L. et al.Pulmonary hypertension associated with benfluorex exposure. Eur Respir J. 2012;40(5):1164–72. [PubMed] [Google Scholar]
- Chin KM, Channick RN, Rubin LJ. Is methamphetamine use associated with idiopathic pulmonary arterial hypertension? Chest. 2006;130(6):1657–63. [PubMed] [Google Scholar]
- Montani D. et al.Pulmonary arterial hypertension in patients treated by dasatinib. Circulation. 2012;125(17):2128–37. [PubMed] [Google Scholar]
- Hachulla E. et al.Early detection of pulmonary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum. 2005;52(12):3792–800. [PubMed] [Google Scholar]
- Mukerjee D. et al.Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: application of a registry approach. Ann Rheum Dis. 2003;62(11):1088–93. [PMC free article] [PubMed] [Google Scholar]
- Launay D. et al.Prevalence and characteristics of moderate to severe pulmonary hypertension in systemic sclerosis with and without interstitial lung disease. J Rheumatol. 2007;34(5):1005–11. [PubMed] [Google Scholar]
- de Groote P. et al.Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann Rheum Dis. 2008;67(1):31–6. [PubMed] [Google Scholar]
- Meune C. et al.Cardiac involvement in systemic sclerosis assessed by tissue-doppler echocardiography during routine care: A controlled study of 100 consecutive patients. Arthritis Rheum. 2008;58(6):1803–9. [PubMed] [Google Scholar]
- Kim KK, Factor SM. Membranoproliferative glomerulonephritis and plexogenic pulmonary arteriopathy in a homosexual man with acquired immunodeficiency syndrome. Hum Pathol. 1987;18(12):1293–6. [PubMed] [Google Scholar]
- Mehta NJ. et al.HIV-Related pulmonary hypertension: analytic review of 131 cases. Chest. 2000;118(4):1133–41. [PubMed] [Google Scholar]
- Opravil M. et al.HIV-associated primary pulmonary hypertension. A case control study. Swiss HIV Cohort Study. Am J Respir Crit Care Med. 1997;155(3):990–5. [PubMed] [Google Scholar]
- Sitbon O. et al.Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviral therapy era. Am J Respir Crit Care Med. 2008;177(1):108–13. [PubMed] [Google Scholar]
- Herve P. et al.Pulmonary vascular disorders in portal hypertension. Eur Respir J. 1998;11(5):1153–66. [PubMed] [Google Scholar]
- Rodriguez-Roisin R. et al.Pulmonary-Hepatic vascular Disorders (PHD) Eur Respir J. 2004;24(5):861–880. [PubMed] [Google Scholar]
- Hadengue A. et al.Pulmonary hypertension complicating portal hypertension: prevalence and relation to splanchnic hemodynamics. Gastroenterology. 1991;100(2):520–8. [PubMed] [Google Scholar]
- Krowka MJ. et al.Portopulmonary hypertension: Results from a 10-year screening algorithm. Hepatology. 2006;44(6):1502–10. [PubMed] [Google Scholar]
- Eisenmenger V. Die angeborene defecte der kammersheidewand des herzen. Z Klin Med. 1897. pp. 132–1.
- Wood P. The Eisenmenger syndrome or pulmonary hypertension with reversed central shunt. Br Med J. 1958;2:701–712. [PMC free article] [PubMed] [Google Scholar]
- Daliento L. et al.Eisenmenger syndrome. Factors relating to deterioration and death. Eur Heart J. 1998;19(12):1845–55. [PubMed] [Google Scholar]
- Besterman E. Atrial Septal Defect with Pulmonary Hypertension. Br Heart J. 1961;23(5):587–598. [PMC free article] [PubMed] [Google Scholar]
- Hoffman JI, Rudolph AM. The natural history of ventricular septal defects in infancy. Am J Cardiol. 1965;16(5):634–53. [PubMed] [Google Scholar]
- Lapa MS. et al.[Clinical characteristics of pulmonary hypertension patients in two reference centers in the city of Sao Paulo] Rev Assoc Med Bras. 2006;52(3):139–43. [PubMed] [Google Scholar]
- Chaves E. The pathology of the arterial pulmonary vasculature in manson’s schistosomiasis. Dis Chest. 1966;50(1):72–7. [PubMed] [Google Scholar]
- de Cleva R. et al.Prevalence of pulmonary hypertension in patients with hepatosplenic Mansonic schistosomiasis–prospective study. Hepatogastroenterology. 2003;50(54):2028–30. [PubMed] [Google Scholar]
- Lapa M. et al.Cardiopulmonary manifestations of hepatosplenic schistosomiasis. Circulation. 2009;119(11):1518–23. [PubMed] [Google Scholar]
- Castro O, Hoque M, Brown BD. Pulmonary hypertension in sickle cell disease: cardiac catheterization results and survival. Blood. 2003;101(4):1257–61. [PubMed] [Google Scholar]
- Gladwin MT. et al.Pulmonary Hypertension as a Risk Factor for Death in Patients with Sickle Cell Disease. N Engl J Med. 2004;350(9):886–895. [PubMed] [Google Scholar]
- Aessopos A. et al.Pulmonary hypertension and right heart failure in patients with beta-thalassemia intermedia. Chest. 1995;107(1):50–3. [PubMed] [Google Scholar]
- Smedema JP, Louw VJ. Pulmonary arterial hypertension after splenectomy for hereditary spherocytosis. Cardiovasc J Afr. 2007;18(2):84–9. [PubMed] [Google Scholar]
- Jais X. et al.An extreme consequence of splenectomy in dehydrated hereditary stomatocytosis: gradual thrombo-embolic pulmonary hypertension and lung-heart transplantation. Hemoglobin. 2003;27(3):139–47. [PubMed] [Google Scholar]
- Stuard ID, Heusinkveld RS, Moss AJ. Microangiopathic hemolytic anemia and thrombocytopenia in primary pulmonary hypertension. N Engl J Med. 1972;287(17):869–70. [PubMed] [Google Scholar]
- Parent F. et al.A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med. 2011;365(1):44–53. [PubMed] [Google Scholar]
- Montani D. et al.Pulmonary veno-occlusive disease: clinical, functional, radiologic, and hemodynamic characteristics and outcome of 24 cases confirmed by histology. Medicine (Baltimore) 2008;87(4):220–33. [PubMed] [Google Scholar]
- Lantuejoul S. et al.Pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis: a clinicopathologic study of 35 cases. Am J Surg Pathol. 2006;30(7):850–7. [PubMed] [Google Scholar]
- Resten A. et al.Pulmonary hypertension: CT of the chest in pulmonary venoocclusive disease. AJR Am J Roentgenol. 2004;183(1):65–70. [PubMed] [Google Scholar]
- Holcomb BW Jr. et al.Pulmonary veno-occlusive disease: a case series and new observations. Chest. 2000;118(6):1671–9. [PubMed] [Google Scholar]
- Dufour B. et al.High-resolution CT of the chest in four patients with pulmonary capillary hemangiomatosis or pulmonary venoocclusive disease. AJR Am J Roentgenol. 1998;171(5):1321–4. [PubMed] [Google Scholar]
- Montani D. et al.Pulmonary veno-occlusive disease. Eur Respir J. 2009;33(1):189–200. [PubMed] [Google Scholar]
- Rabiller A. et al.Occult alveolar haemorrhage in pulmonary veno-occlusive disease. Eur Respir J. 2006;27(1):108–13. [PubMed] [Google Scholar]
- Oudiz RJ. Pulmonary hypertension associated with left-sided heart disease. Clin Chest Med. 2007;28(1):233–41. [PubMed] [Google Scholar]
- Abramson SV. et al.Pulmonary hypertension predicts mortality and morbidity in patients with dilated cardiomyopathy. Ann Intern Med. 1992;116(11):888–95. [PubMed] [Google Scholar]
- Zener JC. et al.Regression of extreme pulmonary hypertension after mitral valve surgery. Am J Cardiol. 1972;30(8):820–6. [PubMed] [Google Scholar]
- Braunwald E. et al.Effects of Mitral-Valve Replacement on the Pulmonary Vascular Dynamics of Patients with Pulmonary Hypertension. N Engl J Med. 1965;273:509–14. [PubMed] [Google Scholar]
- Delgado JF. et al.Pulmonary vascular remodeling in pulmonary hypertension due to chronic heart failure. Eur J Heart Fail. 2005;7(6):1011–6. [PubMed] [Google Scholar]
- Moraes DL, Colucci WS, Givertz MM. Secondary pulmonary hypertension in chronic heart failure: the role of the endothelium in pathophysiology and management. Circulation. 2000;102(14):1718–23. [PubMed] [Google Scholar]
- Fraser KL. et al.Pulmonary hypertension and cardiac function in adult cystic fibrosis: role of hypoxemia. Chest. 1999;115(5):1321–8. [PubMed] [Google Scholar]
- Cottin V. et al.Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005;26(4):586–93. [PubMed] [Google Scholar]
- Weitzenblum E. et al.Prognostic value of pulmonary artery pressure in chronic obstructive pulmonary disease. Thorax. 1981;36(10):752–8. [PMC free article] [PubMed] [Google Scholar]
- Thabut G. et al.Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest. 2005;127(5):1531–6. [PubMed] [Google Scholar]
- Chaouat A. et al.Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172(2):189–94. [PubMed] [Google Scholar]
- Tapson VF, Humbert M. Incidence and prevalence of chronic thromboembolic pulmonary hypertension: from acute to chronic pulmonary embolism. Proc Am Thorac Soc. 2006;3(7):564–7. [PubMed] [Google Scholar]
- Pengo V. et al.Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med. 2004;350(22):2257–64. [PubMed] [Google Scholar]
- Kim NH. Assessment of operability in chronic thromboembolic pulmonary hypertension. Proc Am Thorac Soc. 2006;3(7):584–8. [PubMed] [Google Scholar]
- Dartevelle P. et al.Chronic thromboembolic pulmonary hypertension. Eur Respir J. 2004;23(4):637–48. [PubMed] [Google Scholar]
- Jamieson SW. et al.Pulmonary endarterectomy: experience and lessons learned in 1,500 cases. Ann Thorac Surg. 2003;76(5):1457–62. discussion 1462–4. [PubMed] [Google Scholar]
- Suntharalingam J. et al.Long-term use of sildenafil in inoperable chronic thromboembolic pulmonary hypertension. Chest. 2008;134(2):229–36. [PubMed] [Google Scholar]
- Jais X. et al.Immunosuppressive therapy in lupus- and mixed connective tissue disease-associated pulmonary arterial hypertension: a retrospective analysis of twenty-three cases. Arthritis Rheum. 2008;58(2):521–31. [PubMed] [Google Scholar]
- Rubin LJ. et al.Current and future management of chronic thromboembolic pulmonary hypertension: from diagnosis to treatment responses. Proc Am Thorac Soc. 2006;3(7):601–7. [PubMed] [Google Scholar]
- Dingli D. et al.Unexplained pulmonary hypertension in chronic myeloproliferative disorders. Chest. 2001;120(3):801–8. [PubMed] [Google Scholar]
- Guilpain P. et al.Pulmonary Hypertension Associated with Myeloproliferative Disorders: A Retrospective Study of Ten Cases. Respiration. 2008;76:295–302. [PubMed] [Google Scholar]
- Marvin KS, Spellberg RD. Pulmonary hypertension secondary to thrombocytosis in a patient with myeloid metaplasia. Chest. 1993;103(2):642–4. [PubMed] [Google Scholar]
- Nand S, Orfei E. Pulmonary hypertension in polycythemia vera. Am J Hematol. 1994;47(3):242–4. [PubMed] [Google Scholar]
- Peacock AJ. Pulmonary hypertension after splenectomy: a consequence of loss of the splenic filter or is there something more? Thorax. 2005;60(12):983–4. [PMC free article] [PubMed] [Google Scholar]
- Gluskowski J. et al.Pulmonary haemodynamics at rest and during exercise in patients with sarcoidosis. Respiration. 1984;46(1):26–32. [PubMed] [Google Scholar]
- Bourbonnais JM, Samavati L. Clinical predictors of pulmonary hypertension in sarcoidosis. Eur Respir J. 2008;32(2):296–302. [PubMed] [Google Scholar]
- Handa T. et al.Incidence of pulmonary hypertension and its clinical relevance in patients with sarcoidosis. Chest. 2006;129(5):1246–52. [PubMed] [Google Scholar]
- Nunes H. et al.Pulmonary hypertension associated with sarcoidosis: mechanisms, haemodynamics and prognosis. Thorax. 2006;61(1):68–74. [PMC free article] [PubMed] [Google Scholar]
- Dauriat G. et al.Lung transplantation for pulmonary langerhans’ cell histiocytosis: a multicenter analysis. Transplantation. 2006;81(5):746–50. [PubMed] [Google Scholar]
- Fartoukh M. et al.Severe pulmonary hypertension in histiocytosis X. Am J Respir Crit Care Med. 2000;161(1):216–23. [PubMed] [Google Scholar]
- Harari S. et al.Advanced pulmonary histiocytosis X is associated with severe pulmonary hypertension. Chest. 1997;111(4):1142–4. [PubMed] [Google Scholar]
- Cottin V. et al.Pulmonary hypertension in lymphangioleiomyomatosis: characteristics in 20 patients. Eur Respir J. 2012;40(3):630–40. [PubMed] [Google Scholar]
- Montani D. et al.Pulmonary hypertension in patients with neurofibromatosis type I. Medicine (Baltimore) 2011;90(3):201–11. [PubMed] [Google Scholar]
- Hamaoka K. et al.Pulmonary hypertension in type I glycogen storage disease. Pediatr Cardiol. 1990;11(1):54–6. [PubMed] [Google Scholar]
- Inoue S. et al.[Pulmonary hypertension due to glycogen storage disease type II (Pompe’s disease): a case report] J Cardiol. 1989;19(1):323–32. [PubMed] [Google Scholar]
- Humbert M. et al.Pulmonary arterial hypertension and type-I glycogen-storage disease: the serotonin hypothesis. Eur Respir J. 2002;20(1):59–65. [PubMed] [Google Scholar]
- Pizzo CJ. Type I glycogen storage disease with focal nodular hyperplasia of the liver and vasoconstrictive pulmonary hypertension. Pediatrics. 1980;65(2):341–3. [PubMed] [Google Scholar]
- Elstein D. et al.Echocardiographic assessment of pulmonary hypertension in Gaucher’s disease. Lancet. 1998;351(9115):1544–6. [PubMed] [Google Scholar]
- Lee R, Yousem S. The frequency and type of lung involvement in patients with Gaucher’s disease. Lab Invest. 1998;58:54. A (Abstract) [Google Scholar]
- Theise ND, Scheuer PJ, Grundy JE. Cytomegalovirus and autoimmune liver disease. J Clin Pathol. 1989;42(12):1310–1. [PMC free article] [PubMed] [Google Scholar]
- Chu JW. et al.High prevalence of autoimmune thyroid disease in pulmonary arterial hypertension. Chest. 2002;122(5):1668–73. [PubMed] [Google Scholar]
- Mayer E. et al.Surgical treatment of pulmonary artery sarcoma. J Thorac Cardiovasc Surg. 2001;121(1):77–82. [PubMed] [Google Scholar]
- Anderson MB. et al.Primary pulmonary artery sarcoma: a report of six cases. Ann Thorac Surg. 1995;59(6):1487–90. [PubMed] [Google Scholar]
- Mussot S. et al.Retrospective institutional study of 31 patients treated for pulmonary artery sarcoma. Eur J Cardiothorac Surg. 2013;43(4):787–93. [PubMed] [Google Scholar]
- Yigla M. et al.Pulmonary hypertension in patients with end-stage renal disease. Chest. 2003;123(5):1577–82. [PubMed] [Google Scholar]
- Rich S. et al.Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107(2):216–23. [PubMed] [Google Scholar]
- D’Alonzo GE. et al.Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343–9. [PubMed] [Google Scholar]
- Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351(14):1425–36. [PubMed] [Google Scholar]
- Humbert M. et al.Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173(9):1023–30. [PubMed] [Google Scholar]
- Humbert M. et al.Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122(2):156–63. [PubMed] [Google Scholar]
- Benza RL. et al.Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) Circulation. 2010;122(2):164–72. [PubMed] [Google Scholar]
- Humbert M. et al.Bone morphogenetic protein receptor 2 germline mutations in pulmonary arterial hypertension associated with fenfluramine derivatives. Eur Respir J. 2002;20(3):518–523. [PubMed] [Google Scholar]
- Machado RD. et al.Genetic association of the serotonin transporter in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;173(7):793–7. [PubMed] [Google Scholar]
- Thomson J. et al.Familial and sporadic primary pulmonary hypertension is caused by BMPR2 gene mutations resulting in haploinsufficiency of the bone morphogenetic protein type II receptor. J Heart Lung Transplant. 2001;20(2):149. [PubMed] [Google Scholar]
- Sztrymf B. et al.Clinical outcomes of pulmonary arterial hypertension in carriers of BMPR2 mutation. Am J Respir Crit Care Med. 2008;177(12):1377–83. [PubMed] [Google Scholar]
- Loyd J. et al.Genetic anticipation and abnormal gender ratio at birth in familial primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;152:93–97. [PMC free article] [PubMed] [Google Scholar]
- Loyd JE, Primm RK, Newman JH. Familial primary pulmonary hypertension: clinical patterns. Am Rev Respir Dis. 1984;129(1):194–7. [PubMed] [Google Scholar]
- Deng Z. et al.Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737–744. [PMC free article] [PubMed] [Google Scholar]
- Lane KB. et al.Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet. 2000;26(1):81–4. [PubMed] [Google Scholar]
- Machado RD. et al.Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S32–42. [PMC free article] [PubMed] [Google Scholar]
- Girerd B. et al.Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med. 2010;181(8):851–61. [PubMed] [Google Scholar]
- Rosenzweig EB. et al.Clinical implications of determining BMPR2 mutation status in a large cohort of children and adults with pulmonary arterial hypertension. J Heart Lung Transplant. 2008;27(6):668–74. [PubMed] [Google Scholar]
- Aldred MA. et al.Characterization of the BMPR2 5′-untranslated region and a novel mutation in pulmonary hypertension. Am J Respir Crit Care Med. 2007;176(8):819–24. [PubMed] [Google Scholar]
- Humbert M. Update in pulmonary hypertension 2008. Am J Respir Crit Care Med. 2009. in press. [PubMed]
- Trembath R. et al.Clinical and molecular genetic features of pulmonary hypertension in hereditary hemorrhagic telangiectasia. N Engl J Med. 2001;345:325–334. [PubMed] [Google Scholar]
- Abdalla SA. et al.Primary pulmonary hypertension in families with hereditary haemorrhagic telangiectasia. Eur Respir J. 2004;23(3):373–7. [PubMed] [Google Scholar]
- Harrison RE. et al.Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet. 2003;40(12):865–71. [PMC free article] [PubMed] [Google Scholar]
- Harrison RE. et al.Transforming growth factor-beta receptor mutations and pulmonary arterial hypertension in childhood. Circulation. 2005;111(4):435–41. [PubMed] [Google Scholar]
- Fujiwara M. et al.Implications of mutations of activin receptor-like kinase 1 gene (ALK1) in addition to bone morphogenetic protein receptor II gene (BMPR2) in children with pulmonary arterial hypertension. Circ J. 2008;72(1):127–33. [PubMed] [Google Scholar]
- Smoot LB. et al.Clinical Features of Pulmonary Arterial Hypertension in Young People with an ALK1 Mutation and Hereditary Hemorrhagic Telangiectasia. Arch Dis Child. 2009;94(7):506–11. [PubMed] [Google Scholar]
- Cracowski JL. et al.Évaluation pronostique de biomarqueurs dans l’hypertension artérielle pulmonaire. Rev Mal Respir. 2004;21(6 Pt 1):1137–43. [PubMed] [Google Scholar]
- Morrell N. et al.Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta1 and bone morphogenetic proteins. Circulation. 2001;104:790–795. [PubMed] [Google Scholar]
- Masri F. et al.Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293(3):L548–54. [PubMed] [Google Scholar]
- Sztrymf B. et al.Genes and pulmonary arterial hypertension. Respiration. 2007;74(2):123–32. [PubMed] [Google Scholar]
- Girerd B. et al.Absence of influence of gender and BMPR2 mutation type on clinical phenotypes of pulmonary arterial hypertension. Respir Res. 2010;11:73. [PMC free article] [PubMed] [Google Scholar]
- West J. et al.Gene expression in BMPR2 mutation carriers with and without evidence of pulmonary arterial hypertension suggests pathways relevant to disease penetrance. BMC Med Genomics. 2008;1:45. [PMC free article] [PubMed] [Google Scholar]
- Austin ED. et al.Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir J. 2009;34(5):1093–9. [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan DS. et al.Treatment of pulmonary arterial hypertension with targeted therapies. Nat Rev Cardiol. 2011;8(9):526–38. [PubMed] [Google Scholar]
- Stenmark KR. et al.Hypoxic activation of adventitial fibroblasts: role in vascular remodeling. Chest. 2002;122(6 Suppl):326S–334S. [PubMed] [Google Scholar]
- Davie NJ. et al.Hypoxia-induced pulmonary artery adventitial remodeling and neovascularization: contribution of progenitor cells. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L668–78. [PubMed] [Google Scholar]
- Cool CD. et al.Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell-specific markers. Evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am J Pathol. 1999;155(2):411–9. [PMC free article] [PubMed] [Google Scholar]
- Montani D. et al.C-Kit Positive Cells Accumulate in Remodeled Vessels of Idiopathic Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2011;184(1):116–23. [PubMed] [Google Scholar]
- Gambaryan N. et al.Circulating fibrocytes and pulmonary arterial hypertension. Eur Respir J. 2012;39(1):210–2. [PubMed] [Google Scholar]
- Gambaryan N. et al.Targeting of c-kit + haematopoietic progenitor cells prevents hypoxic pulmonary hypertension. Eur Respir J. 2011;37(6):1392–9. [PubMed] [Google Scholar]
- Kherbeck N. et al.The Role of Inflammation and Autoimmunity in the Pathophysiology of Pulmonary Arterial Hypertension. Clin Rev Allergy Immunol. 2013;44(1):31–8. [PubMed] [Google Scholar]
- Balabanian K. et al.CX(3)C chemokine fractalkine in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2002;165(10):1419–25. [PubMed] [Google Scholar]
- Itoh T. et al.Increased plasma monocyte chemoattractant protein-1 level in idiopathic pulmonary arterial hypertension. Respirology. 2006;11(2):158–63. [PubMed] [Google Scholar]
- Nicolls MR. et al.Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J. 2005;26(6):1110–8. [PubMed] [Google Scholar]
- Perros F. et al.Dendritic cell recruitment in lesions of human and experimental pulmonary hypertension. Eur Respir J. 2007;29(3):462–8. [PubMed] [Google Scholar]
- Herve P. et al.Pathobiology of pulmonary hypertension. The role of platelets and thrombosis. Clin Chest Med. 2001;22(3):451–8. [PubMed] [Google Scholar]
- Herve P. et al.Increased plasma serotonin in primary pulmonary hypertension. Am J Med. 1995;99:249–254. [PubMed] [Google Scholar]
- Huertas A. et al.Leptin and regulatory T lymphocytes in idiopathic pulmonary arterial hypertension. Eur Respir J. 2012;40(4):895–904. [PubMed] [Google Scholar]
- Perros F. et al.Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185(3):311–21. [PubMed] [Google Scholar]
- Humbert M. et al.Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):13S–24S. [PubMed] [Google Scholar]
- Christman BW. et al.An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327(2):70–5. [PubMed] [Google Scholar]
- Montani D. et al.Phosphodiesterase type 5 inhibitors in pulmonary arterial hypertension. Adv Ther. 2009;26(9):813–25. [PubMed] [Google Scholar]
- Petkov V. et al.Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest. 2003;111(9):1339–46. [PMC free article] [PubMed] [Google Scholar]
- Jeffery T, Morrell NW. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog Cardiovasc Dis. 2002;45:173–202. [PubMed] [Google Scholar]
- Giaid A. et al.Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328(24):1732–9. [PubMed] [Google Scholar]
- Yuan XJ. et al.Attenuated K + channel gene transcription in primary pulmonary hypertension. Lancet. 1998;351(9104):726–7. [PubMed] [Google Scholar]
- Yuan JX. et al.Dysfunctional voltage-gated K + channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation. 1998;98(14):1400–6. [PubMed] [Google Scholar]
- Weir EK. et al.Anorexic Agents Aminorex, Fenfluramine, and Dexfenfluramine Inhibit Potassium Current in Rat Pulmonary Vascular Smooth Muscle and Cause Pulmonary Vasoconstriction. Circulation. 1996;94(9):2216–2220. [PubMed] [Google Scholar]
- MacLean MR. et al.5-hydroxytryptamine and the pulmonary circulation: receptors, transporters and relevance to pulmonary arterial hypertension. Br J Pharmacol. 2000;131(2):161–8. [PMC free article] [PubMed] [Google Scholar]
- Marcos E. et al.Serotonin transporter inhibitors protect against hypoxic pulmonary hypertension. Am J Respir Crit Care Med. 2003;168(4):487–93. [PubMed] [Google Scholar]
- Keegan A. et al.Contribution of the 5-HT(1B) receptor to hypoxia-induced pulmonary hypertension: converging evidence using 5-HT(1B)-receptor knockout mice and the 5-HT(1B/1D)-receptor antagonist GR127935. Circ Res. 2001;89(12):1231–9. [PubMed] [Google Scholar]
- Guilluy C. et al.Inhibition of RhoA/Rho kinase pathway is involved in the beneficial effect of sildenafil on pulmonary hypertension. Br J Pharmacol. 2005;146(7):1010–8. [PMC free article] [PubMed] [Google Scholar]
- Nagaoka T. et al.Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med. 2005;171(5):494–9. [PubMed] [Google Scholar]
- Guilluy C. et al.RhoA and Rho kinase activation in human pulmonary hypertension: role of 5-HT signaling. Am J Respir Crit Care Med. 2009;179(12):1151–8. [PubMed] [Google Scholar]
- Semenza GL. HIF-1 and mechanisms of hypoxia sensing. Curr Opin Cell Biol. 2001;13(2):167–71. [PubMed] [Google Scholar]
- Semenza GL. Involvement of hypoxia-inducible factor 1 in pulmonary pathophysiology. Chest. 2005;128(6 Suppl):592S–594S. [PubMed] [Google Scholar]
- Tuder RM. et al.Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol. 2001;195(3):367–74. [PubMed] [Google Scholar]
- Remillard CV, Yuan JX. High altitude pulmonary hypertension: role of K + and Ca2+ channels. High Alt Med Biol. 2005;6(2):133–46. [PMC free article] [PubMed] [Google Scholar]
- Cool CD. et al.Pathogenesis and evolution of plexiform lesions in pulmonary hypertension associated with scleroderma and human immunodeficiency virus infection. Hum Pathol. 1997;28(4):434–42. [PubMed] [Google Scholar]
- Pietra GG. et al.Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):25S–32S. [PubMed] [Google Scholar]
- Bjornsson J, Edwards WD. Primary pulmonary hypertension: a histopathologic study of 80 cases. Mayo Clin Proc. 1985;60(1):16–25. [PubMed] [Google Scholar]
- Heath D, Edwards JE. The pathology of hypertensive pulmonary vascular disease; a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation. 1958;18(4 Part 1):533–47. [PubMed] [Google Scholar]
- Dorfmuller P. et al.Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22(2):358–63. [PubMed] [Google Scholar]
- Dorfmuller P. et al.Chemokine RANTES in severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2002;165(4):534–9. [PubMed] [Google Scholar]
- Dorfmuller P. et al.Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum Pathol. 2007;38(6):893–902. [PubMed] [Google Scholar]
- Overbeek MJ. et al.Pulmonary arterial hypertension in limited cutaneous systemic sclerosis: a distinctive vasculopathy. Eur Respir J. 2009;34(2):371–9. [PubMed] [Google Scholar]
- Golde DW. et al.Occult pulmonary haemorrhage in leukaemia. Br Med J. 1975;2(5964):166–8. [PMC free article] [PubMed] [Google Scholar]
- Tron V. et al.Pulmonary capillary hemangiomatosis. Hum Pathol. 1986;17(11):1144–50. [PubMed] [Google Scholar]
- Runo JR. et al.Pulmonary veno-occlusive disease caused by an inherited mutation in bone morphogenetic protein receptor II. Am J Respir Crit Care Med. 2003;167(6):889–94. [PubMed] [Google Scholar]
- Barst RJ. et al.Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):40S–47S. [PubMed] [Google Scholar]
- Elliott CG. et al.Pulmonary veno-occlusive disease associated with severe reduction of single-breath carbon monoxide diffusing capacity. Respiration. 1988;53(4):262–6. [PubMed] [Google Scholar]
- Groepenhoff H. et al.Exercise testing to estimate survival in pulmonary hypertension. Med Sci Sports Exerc. 2008;40(10):1725–32. [PubMed] [Google Scholar]
- Wensel R. et al.Assessment of survival in patients with primary pulmonary hypertension: importance of cardiopulmonary exercise testing. Circulation. 2002;106(3):319–24. [PubMed] [Google Scholar]
- McQuillan BM. et al.Clinical correlates and reference intervals for pulmonary artery systolic pressure among echocardiographically normal subjects. Circulation. 2001;104(23):2797–802. [PubMed] [Google Scholar]
- Tunariu N. et al.Ventilation-perfusion scintigraphy is more sensitive than multidetector CTPA in detecting chronic thromboembolic pulmonary disease as a treatable cause of pulmonary hypertension. J Nucl Med. 2007;48(5):680–4. [PubMed] [Google Scholar]
- Ley S. et al.Diagnostic performance of state-of-the-art imaging techniques for morphological assessment of vascular abnormalities in patients with chronic thromboembolic pulmonary hypertension (CTEPH) Eur Radiol. 2012;22(3):607–16. [PubMed] [Google Scholar]
- Marcus JT. et al.Interventricular mechanical asynchrony in pulmonary arterial hypertension: left-to-right delay in peak shortening is related to right ventricular overload and left ventricular underfilling. J Am Coll Cardiol. 2008;51(7):750–7. [PubMed] [Google Scholar]
- Haddad F. et al.Right ventricular function in cardiovascular disease, part I: Anatomy, physiology, aging, and functional assessment of the right ventricle. Circulation. 2008;117(11):1436–48. [PubMed] [Google Scholar]
- Galie N. et al.Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT) Eur Heart J. 2009;30(20):2493–537. [PubMed] [Google Scholar]
- van Wolferen SA. et al.Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J. 2007;28(10):1250–7. [PubMed] [Google Scholar]
- Naeije R. Hepatopulmonary syndrome and portopulmonary hypertension. Swiss Med Wkly. 2003;133(11–12):163–9. [PubMed] [Google Scholar]
- Hoeper MM. et al.Complications of right heart catheterization procedures in patients with pulmonary hypertension in experienced centers. J Am Coll Cardiol. 2006;48(12):2546–52. [PubMed] [Google Scholar]
- Benza RL. et al.The REVEAL Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest. 2012;141(2):354–62. [PubMed] [Google Scholar]
- Sitbon O, Humbert M, Simonneau G. Primary pulmonary hypertension: Current therapy. Prog Cardiovasc Dis. 2002;45(2):115–28. [PubMed] [Google Scholar]
- Bonnin M. et al.Severe pulmonary hypertension during pregnancy: mode of delivery and anesthetic management of 15 consecutive cases. Anesthesiology. 2005;102(6):1133–7. discussion 5A-6A. [PubMed] [Google Scholar]
- Price LC. et al.Non-cardiothoracic non-obstetric surgery in mild-moderate pulmonary hypertension: Perioperative management of 28 consecutive individual cases. Eur Respir J. 2010;35(6):1294–302. [PubMed] [Google Scholar]
- Provencher S. et al.Heart rate responses during the 6-minute walk test in pulmonary arterial hypertension. Eur Respir J. 2006;27(1):114–20. [PubMed] [Google Scholar]
- Sitbon O. et al.Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol. 2002;40(4):780–8. [PubMed] [Google Scholar]
- Fuster V. et al.Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation. 1984;70(4):580–7. [PubMed] [Google Scholar]
- Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327(2):76–81. [PubMed] [Google Scholar]
- Rich S. et al.The short-term effects of digoxin in patients with right ventricular dysfunction from pulmonary hypertension. Chest. 1998;114(3):787–92. [PubMed] [Google Scholar]
- Sitbon O. et al.Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111(23):3105–11. [PubMed] [Google Scholar]
- Moncada S. et al.An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. 1976;263(5579):663–5. [PubMed] [Google Scholar]
- Rubin LJ. et al.Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann Intern Med. 1990;112(7):485–91. [PubMed] [Google Scholar]
- O’Callaghan DS. et al.Treatment of pulmonary arterial hypertension with targeted therapies. Nat Rev Cardiol. 2011;8(9):526–38. [PubMed] [Google Scholar]
- Higenbottam T. et al.Long-term treatment of primary pulmonary hypertension with continuous intravenous epoprostenol (prostacyclin) Lancet. 1984;1(8385):1046–7. [PubMed] [Google Scholar]
- Robbins IM. et al.A survey of diagnostic practices and the use of epoprostenol in patients with primary pulmonary hypertension. Chest. 1998;114(5):1269–75. [PubMed] [Google Scholar]
- Barst RJ. et al.A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996;334(5):296–302. [PubMed] [Google Scholar]
- Badesch DB. et al.Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med. 2000;132(6):425–34. [PubMed] [Google Scholar]
- Barst RJ. et al.Survival in primary pulmonary hypertension with long-term continuous intravenous prostacyclin. Ann Intern Med. 1994;121(6):409–15. [PubMed] [Google Scholar]
- McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation. 2002;106(12):1477–82. [PubMed] [Google Scholar]
- Rosenzweig EB, Kerstein D, Barst RJ. Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation. 1999;99(14):1858–65. [PubMed] [Google Scholar]
- Nunes H. et al.Prognostic factors for survival in human immunodeficiency virus-associated pulmonary arterial hypertension. Am J Respir Crit Care Med. 2003;167(10):1433–9. [PubMed] [Google Scholar]
- Cabrol S. et al.Intravenous epoprostenol in inoperable chronic thromboembolic pulmonary hypertension. J Heart Lung Transplant. 2007;26(4):357–62. [PubMed] [Google Scholar]
- McLaughlin VV. et al.Reduction in pulmonary vascular resistance with long-term epoprostenol (prostacyclin) therapy in primary pulmonary hypertension. N Engl J Med. 1998;338(5):273–7. [PubMed] [Google Scholar]
- Shapiro SM. et al.Primary pulmonary hypertension: improved long-term effects and survival with continuous intravenous epoprostenol infusion. J Am Coll Cardiol. 1997;30(2):343–9. [PubMed] [Google Scholar]
- Laliberte K. et al.Pharmacokinetics and steady-state bioequivalence of treprostinil sodium (Remodulin) administered by the intravenous and subcutaneous route to normal volunteers. J Cardiovasc Pharmacol. 2004;44(2):209–14. [PubMed] [Google Scholar]
- Simonneau G. et al.Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(6):800–4. [PubMed] [Google Scholar]
- McLaughlin VV. et al.Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol. 2010;55(18):1915–22. [PubMed] [Google Scholar]
- Olschewski H. et al.Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347(5):322–9. [PubMed] [Google Scholar]
- Hoeper MM. et al.Long-term treatment of primary pulmonary hypertension with aerosolized iloprost, a prostacyclin analogue. N Engl J Med. 2000;342(25):1866–70. [PubMed] [Google Scholar]
- Channick RN. et al.Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 2001;358(9288):1119–23. [PubMed] [Google Scholar]
- Rubin LJ. et al.Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346(12):896–903. [PubMed] [Google Scholar]
- Humbert M. et al.Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J. 2004;24(3):353–9. [PubMed] [Google Scholar]
- Galie N. et al.Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet. 2008;371(9630):2093–100. [PubMed] [Google Scholar]
- Sitbon O. et al.Effects of the dual endothelin receptor antagonist bosentan in patients with pulmonary arterial hypertension: a 1-year follow-up study. Chest. 2003;124(1):247–54. [PubMed] [Google Scholar]
- McLaughlin VV. et al.Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J. 2005;25(2):244–9. [PubMed] [Google Scholar]
- Sitbon O. et al.Survival in patients with class III idiopathic pulmonary arterial hypertension treated with first line oral bosentan compared with an historical cohort of patients started on intravenous epoprostenol. Thorax. 2005;60(12):1025–30. [PMC free article] [PubMed] [Google Scholar]
- Galie N. et al.Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117(23):3010–9. [PubMed] [Google Scholar]
- Klinger JR. et al.Long-term pulmonary hemodynamic effects of ambrisentan in pulmonary arterial hypertension. Am J Cardiol. 2011;108(2):302–7. [PubMed] [Google Scholar]
- Ghofrani HA. et al.Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet. 2002;360(9337):895–900. [PubMed] [Google Scholar]
- Schermuly RT. et al.Chronic sildenafil treatment inhibits monocrotaline-induced pulmonary hypertension in rats. Am J Respir Crit Care Med. 2004;169(1):39–45. [PubMed] [Google Scholar]
- Galie N. et al.Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148–57. [PubMed] [Google Scholar]
- Galie N. et al.Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119(22):2894–903. [PubMed] [Google Scholar]
- Barst RJ. et al.Tadalafil monotherapy and as add-on to background bosentan in patients with pulmonary arterial hypertension. J Heart Lung Transplant. 2011;30(6):632–43. [PubMed] [Google Scholar]
- Simonneau G. et al.Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med. 2008;149(8):521–30. [PubMed] [Google Scholar]
- McLaughlin VV. et al.Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2006;174(11):1257–63. [PubMed] [Google Scholar]
- Hoeper MM. et al.Bosentan treatment in patients with primary pulmonary hypertension receiving nonparenteral prostanoids. Eur Respir J. 2003;22(2):330–4. [PubMed] [Google Scholar]
- Ghofrani HA. et al.Oral sildenafil as long-term adjunct therapy to inhaled iloprost in severe pulmonary arterial hypertension. J Am Coll Cardiol. 2003;42(1):158–64. [PubMed] [Google Scholar]
- Gomberg-Maitland M. et al.Efficacy and safety of sildenafil added to treprostinil in pulmonary hypertension. Am J Cardiol. 2005;96(9):1334–6. [PubMed] [Google Scholar]
- Hoeper MM. et al.Combination therapy with bosentan and sildenafil in idiopathic pulmonary arterial hypertension. Eur Respir J. 2004;24(6):1007–10. [PubMed] [Google Scholar]
- Mathai SC. et al.Addition of sildenafil to bosentan monotherapy in pulmonary arterial hypertension. Eur Respir J. 2007;29(3):469–75. [PubMed] [Google Scholar]
- Rubin L. et al.Effect of Macitentan on Morbidity and Mortality in Pulmonary Arterial Hypertension (PAH): Results From the SERAPHIN Trial. Chest. 2012;142:1026A–1026A. doi: 10.1378/chest.1456207. [CrossRef] [Google Scholar]
- Iglarz M. et al.Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. J Pharmacol Exp Ther. 2008;327(3):736–45. [PubMed] [Google Scholar]
- Sidharta PN. et al.Macitentan: entry-into-humans study with a new endothelin receptor antagonist. Eur J Clin Pharmacol. 2011;67(10):977–84. [PMC free article] [PubMed] [Google Scholar]
- Jing ZC. et al.Vardenafil in pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled study. Am J Respir Crit Care Med. 2011;183(12):1723–9. [PubMed] [Google Scholar]
- Ghofrani HA, Grimminger F. Soluble guanylate cyclase stimulation: an emerging option in pulmonary hypertension therapy. Eur Respir Rev. 2009;18(111):35–41. [PubMed] [Google Scholar]
- Grimminger F. et al.First acute haemodynamic study of soluble guanylate cyclase stimulator riociguat in pulmonary hypertension. Eur Respir J. 2009;33(4):785–92. [PubMed] [Google Scholar]
- Ghofrani HA. et al.Riociguat for chronic thromboembolic pulmonary hypertension and pulmonary arterial hypertension: a phase II study. Eur Respir J. 2010;36(4):792–9. [PubMed] [Google Scholar]
- Schermuly RT. et al.Riociguat for the treatment of pulmonary hypertension. Expert Opin Investig Drugs. 2011;20(4):567–76. [PubMed] [Google Scholar]
- Ghio S. et al.Left ventricular systolic dysfunction associated with pulmonary hypertension riociguat trial (LEPHT): rationale and design. Eur J Heart Fail. 2012;14(8):946–53. [PubMed] [Google Scholar]
- Hoeper MM. et al.Riociguat for interstitial lung disease and pulmonary hypertension: a pilot trial. Eur Respir J. 2013;41(4):853–60. [PubMed] [Google Scholar]
- Ghofrani H. et al.Riociguat for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med . 2013;369(4):330–40. [PubMed] [Google Scholar]
- Asaki T. et al.Structure-activity studies on diphenylpyrazine derivatives: a novel class of prostacyclin receptor agonists. Bioorg Med Chem. 2007;15(21):6692–704. [PubMed] [Google Scholar]
- Kuwano K. et al.2-[4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy]-N-(methylsulfonyl)acetam ide (NS-304), an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther. 2007;322(3):1181–8. [PubMed] [Google Scholar]
- Kuwano K. et al.A long-acting and highly selective prostacyclin receptor agonist prodrug, 2-{4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}-N-(methylsulfonyl)acetam ide (NS-304), ameliorates rat pulmonary hypertension with unique relaxant responses of its active form, {4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}acetic acid (MRE-269), on rat pulmonary artery. J Pharmacol Exp Ther. 2008;326(3):691–9. [PubMed] [Google Scholar]
- Morrison K. et al.Differential effects of selexipag and prostacyclin analogs in rat pulmonary artery. J Pharmacol Exp Ther. 2012;343(3):547–55. [PubMed] [Google Scholar]
- Simonneau G. et al.Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J. 2012;40(4):874–80. [PubMed] [Google Scholar]
- NIH. ClinicalTrials.gov. ACT-BR;293987 in pulmonary arterial hypertension [online] 2011.
- Barst RJ. PDGF signaling in pulmonary arterial hypertension. J Clin Invest. 2005;115(10):2691–4. [PMC free article] [PubMed] [Google Scholar]
- Perros F. et al.Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178(1):81–8. [PubMed] [Google Scholar]
- Panos RJ, Baker SK. Mediators, cytokines, and growth factors in liver-lung interactions. Clin Chest Med. 1996;17(1):151–69. [PubMed] [Google Scholar]
- Schermuly RT. et al.Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115(10):2811–21. [PMC free article] [PubMed] [Google Scholar]
- Ghofrani H. et al.Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am J Respir Crit Care Med. 2010;182(9):1171–7. [PMC free article] [PubMed] [Google Scholar]
- Hoeper MM. et al.Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation. 2013;127(10):1128–38. [PubMed] [Google Scholar]
- Kerkela R. et al.Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. 2006;12(8):908–16. [PubMed] [Google Scholar]
- McLaughlin VV. et al.Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(1 Suppl):78S–92S. [PubMed] [Google Scholar]
- Sandoval J. et al.Graded balloon dilation atrial septostomy in severe primary pulmonary hypertension. A therapeutic alternative for patients nonresponsive to vasodilator treatment. J Am Coll Cardiol. 1998;32(2):297–304. [PubMed] [Google Scholar]
- Rothman A. et al.Atrial septostomy as a bridge to lung transplantation in patients with severe pulmonary hypertension. Am J Cardiol. 1999;84(6):682–6. [PubMed] [Google Scholar]
- Kerstein D. et al.Blade balloon atrial septostomy in patients with severe primary pulmonary hypertension. Circulation. 1995;91(7):2028–35. [PubMed] [Google Scholar]
- Kurzyna M. et al.Atrial septostomy in treatment of end-stage right heart failure in patients with pulmonary hypertension. Chest. 2007;131(4):977–83. [PubMed] [Google Scholar]
- Hosenpud JD. et al.The Registry of the International Society for Heart and Lung Transplantation: eighteenth Official Report-2001. J Heart Lung Transplant. 2001;20(8):805–15. [PubMed] [Google Scholar]
- Levine SM. et al.Single lung transplantation for primary pulmonary hypertension. Chest. 1990;98(5):1107–15. [PubMed] [Google Scholar]
- Pielsticker EJ, Martinez FJ, Rubenfire M. Lung and heart-lung transplant practice patterns in pulmonary hypertension centers. J Heart Lung Transplant. 2001;20(12):1297–304. [PubMed] [Google Scholar]
- Mendeloff EN. et al.Lung transplantation for pulmonary vascular disease. Ann Thorac Surg. 2002;73(1):209–17. discussion 217–9. [PubMed] [Google Scholar]
- Reitz BA. et al.Heart-lung transplantation: successful therapy for patients with pulmonary vascular disease. N Engl J Med. 1982;306(10):557–64. [PubMed] [Google Scholar]