An Update on the Pathophysiology and Treatment of Cardiorenal Syndrome
Abstract
Cardiorenal syndrome (CRS) encompasses various disorders of the heart and kidneys; dysfunction of one organ leads to acute or chronic dysfunction of the other. It incorporates the intersection of heart-kidney interactions across several mediums, hemodynamically, through the alterations in neurohormonal markers, and increased venous and renal pressure, all of which are hallmarks of its clinical phenotypes. This article explores the epidemiology, pathology, classification and treatment of each type of CRS.
Introduction
Cardiac and renal diseases share common vascular risk factors, such as hypertension and diabetes. The overlapping risk factors can be observed in various hemodynamic interactions in heart and kidney failure, neurohormonal interplay, chronic kidney disease (CKD) and atherosclerotic diseases [1]. Concurrent renal disease and heart failure (HF) are directly related and indicative of a worse prognosis leading to a greater burden on our healthcare system and patient quality of life [2]. A 2008 consensus conference defined “disorders of the heart and kidneys whereby acute or chronic dysfunction in one organ may induce acute or chronic dysfunction of the other” as cardiorenal syndrome (CRS) [3]. This review will describe the pathophysiology in CRS, classified subtypes, general risk factors and diagnosis techniques, and treatment and management of the various symptoms that arise from this condition. An outline of each subtype is illustrated in Table 1. The information presented serves as an update to Vinod et al’s “role of arginine vasopressin and vaptans in heart failure”. This manuscript will not only detail the use of specific treatment option but will dive into all the common treatments across all types of CRS.
Table 1
| Cardiorenal types | Characteristics | Causes of morbidity |
|---|---|---|
| Type 1 (acute cardiorenal) | Acute cardiac impairment leading to acute kidney injury (AKI) | Cardiogenic shock and AKI, acute decompensated heart failure (ADHF) resulting in AKI |
| Type 2 (chronic cardiorenal) | Chronic cardiac impairment leading to renal impairment | Chronic heart failure |
| Type 3 (acute renocardiac) | AKI leading to cardiac impairment | Heart failure in the setting of AKI from volume overload, inflammatory surge and accompanying metabolic disturbances |
| Type 4 (chronic renocardiac) | Chronic kidney disease (CKD) leading to cardiac impairment | Myocardial remodeling and heart failure from CKD-associated cardiomyopathy |
| Type 5 (secondary cardiorenal) | Systemic condition leading to both cardiac and renal impairment | Diabetes, amyloidosis and sepsis |
Methodology
This article serves to provide a current overview of the classifications, pathology, risk factors, diagnosis and management of CRS. The authors used MEDLINE and Cochrane Central Register of Controlled Trials to order, identify and analyze the latest information available. Bibliography searches of primary articles were used as well. Important Medical Subject Heading descriptors used for the search included cardiorenal syndrome, kidney disease, chronic renal/chronic kidney, acute kidney injury, end-stage renal or end-stage kidney disease, renal dysfunction, heart failure, ultrafiltration, ACE inhibitor ARB MRA, beta blockers, cardiac resynchronization therapy, LVAD, diuretics and loop diuretic. All authors assisted in the collection, writing and reviewing of the manuscript presented.
Discussion
Pathophysiology
Increased venous pressure
In order to oxygenate the kidney, the difference between the arterial driving pressure and venous outflow pressures must remain large enough to allow for adequate renal blood flow and glomerular filtration rate [1]. In HF, increased central venous pressure (CVP) leads to a reduction of both perfusion gradient across the glomerular capillary bed and renal function [4, 5]. Right ventricular dilatation and dysfunction can lead to elevated venous pressure and can impair left ventricular (LV) filling and reduce cardiac output. Elevated blood pressure and central venous congestion directly impact decreasing renal function for patients with acute heart failure (AHF) [6]. Elevated CVP results in renal venous hypertension and increased renal resistance, and impaired intrarenal blood flow was present in patients with AHF. Seventy-five percent of the patients who presented with a baseline CVP > 24 mm Hg developed worsening renal function (WRF) according to Mullens et al [6]. Reduction in blood flow can be present in patients despite the preservation of glomerular filtration rate (GFR). Patients with severe decompensated heart failure display both decreased renal blood flow and increased venous pressure, causing declining GFR and decreased filtration fraction [7]. Increased CVP has a detrimental effect on renal function due to continuous activation of renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system (SNS) pathway.
Neurohormonal system
Neurohormonal systems such as the RAAS and the SNS are homeostatic systems responsible for proper blood flow during periods of hypotension. Activation of RAAS occurs in response to decreased cardiac output or decreased perfusion within the renal system [4]. RAAS leads to increased production of angiotensin II, which causes systemic vasoconstriction and extracellular volume expansion via sodium retention. While normal filtration of the kidney is maintained, overactivation can lead to the glomerular system damage and worsened heart congestion [7, 8]. Decreased renal function is strongly correlated with changes in systemic blood pressure, which can be the strongest hemodynamic driver for decreasing renal function in humans [9]. The neurohormonal pathways often lead to injury and worse clinical outcomes due to their activation of oxidative and inflammatory pathways. RAAS and SNS activation increase the presence of inflammatory molecules in the kidney when present in HF along with cardiac remodeling at the ultrastructural and molecular levels. In RAAS, renin and inflammatory markers are commonly upregulated [10]. Angiotensin II binding to AT1R and AT2R receptors can lead to the activation of reactive oxidation species pathways in the body. These reactive oxygen species (ROS) can cause arrhythmias, hypertrophy, hypertension and cell death [11].
Classification
CRS type 1 (acute CRS)
CRS type 1 is characterized by acute (or acute on chronic) congestive heart failure resulting in acute kidney injury (AKI), with mechanisms of progression illustrated in Figure 1 [12]. Approximately 25% of patients with acute decompensated heart failure (ADHF) develop AKI [13]. Acute congestive heart failure’s effect on the renal system remains the same, as water and sodium reabsorption increases despite increases in extracellular fluid volumes, which contribute to fluid overload [14]. Hemodynamically, there are multiple mechanisms that derive from ADHF which produce decreased arterial flow and GFR. RAAS and SNS activation can cause efferent vasoconstriction and decreased blood flow and extracellular fluid. High CVP can directly affect the perfusion pressure of the kidney and renal veins as well as produce tubular collapse and declining GFR [12].
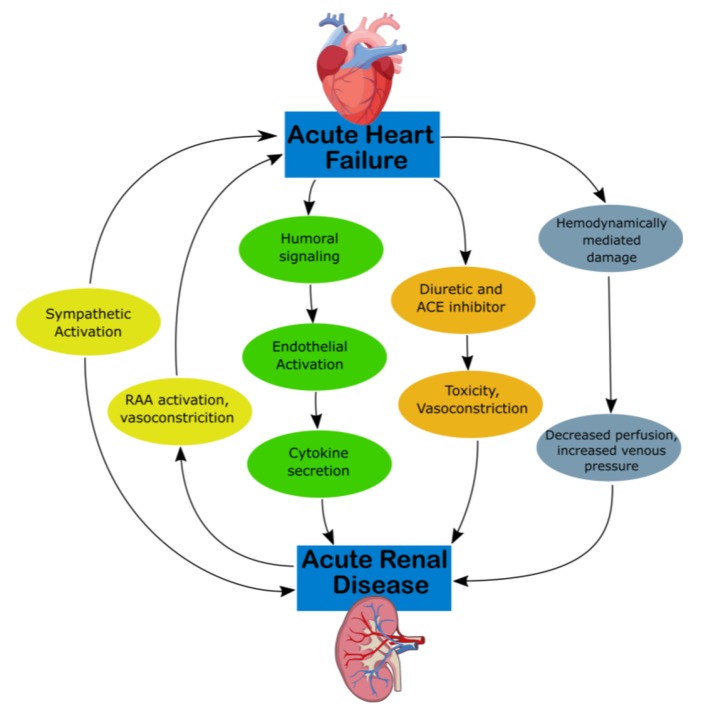
Mechanism of damage in CRS-1. ACE, angiotensin-converting enzyme; CKD, chronic kidney disease; CRS, cardiorenal syndrome; RAA, rennin-angiotensin-aldosterone.
CRS type 2 (chronic CRS)
CRS-2 is characterized by chronic cardiac problems leading to progressive CKD. The mechanisms of progression can be seen in Figure 2. CKD is especially detrimental in patients with HF, resulting in prolonged hospitalization and worse clinical outcomes [13]. The renal response to chronic heart failure (CHF) presents as low renal plasma flow and relative preservation of GFR. The GFR remains stable until the continual increase in efferent arteriolar resistance and glomerular capillary hydrostatic pressure causes severe cardiac injury [12]. HF with preserved and reduced ejection fraction (EF), ischemic cardiomyopathy, atrial fibrillation (AF) and congenital heart disease are among the underlying conditions of CRS-2. To be categorized as CRS-2, CKD must present secondarily to CHF [13]. The combination of renal congestion and hypoperfusion creates increased pressure in the right atrium which influences renal dysfunction for CHF patients [12]. CKD is often associated with erythropoietin deficiency. Pallazzuoli et al found that erythropoiesis stimulating agents can be used to treat HF, CKD and anemia with positive increases in cardiac functions and reduced left ventricle size and volume [14]. The ADHERE Study examined the importance of CKD in HF patients involving 118,465 patients with decompensated HF. The vast majority, 91%, had impaired kidney function, while 56% had an estimated GFR (eGFR) of 15 – 69 mL/min/m2. While worse outcomes are associated with low GFR, the relationship between GFR and LVEF remains unknown. CHF patients have similar GFR for both impaired and preserved LV function [13]. Furthermore, RAAS overactivation and a lack of a subsequent decrease in circulating volume can damage the body. RAAS activation leads to the progression of CKD through the damage of glomerular filtration units [12].
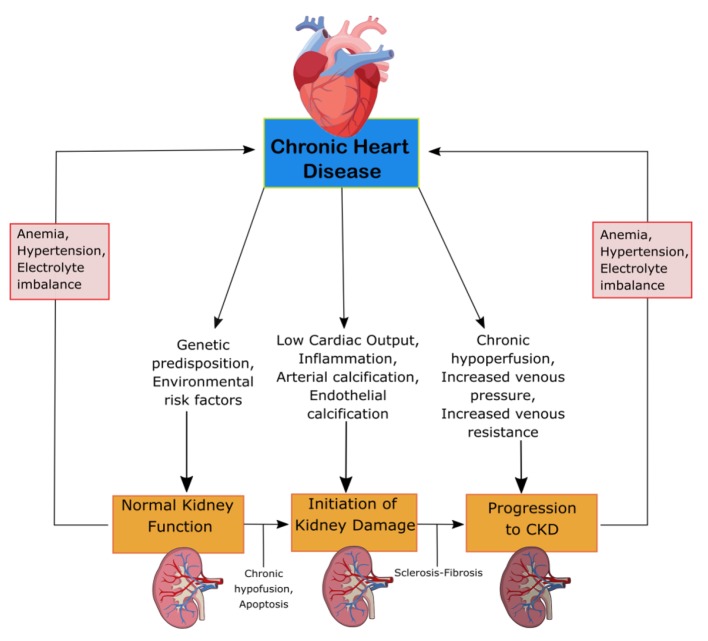
Mechanism of damage in CRS-2. CKD, chronic kidney disease; CRS, cardiorenal syndrome.
CRS type 3 (acute renocardiac syndrome)
CRS-3 is identified by an abruptly decreased kidney function secondary to AKI, ischemia, or glomerulonephritis producing acute cardiac dysfunction [12]. The mechanisms of progression are identified in Figure 3. Identifying this subtype is complex due to differing methods in defining AKI and determining the incidence of acute cardiac dysfunction [5]. It is difficult to establish causality of cardiac disease due to AKI given that multiple comorbid conditions can predispose to AKI. Thus, kidney injury is usually secondary to multiorgan failure of various cases [15]. AKI, however, is associated with an 86% increased risk of cardiovascular mortality and a 38% increased risk of major cardiovascular events [16]. Multiple pathways have been identified for how AKI can affect heart function. Fluid overload can lead to the development of pulmonary edema and increased mortality. These patients are often at higher risk for needing renal replacement therapy [4]. AKI can lead to the development of hyperkalemia, disrupting electrolyte balance and causing arrhythmias, resulting in cardiac arrest. Early stages of hyperkalemia can be diagnosed by a narrowing and peaking of the T wave on electrocardiogram (EKG), which correlates with increased activity of potassium channels and faster repolarization rates.
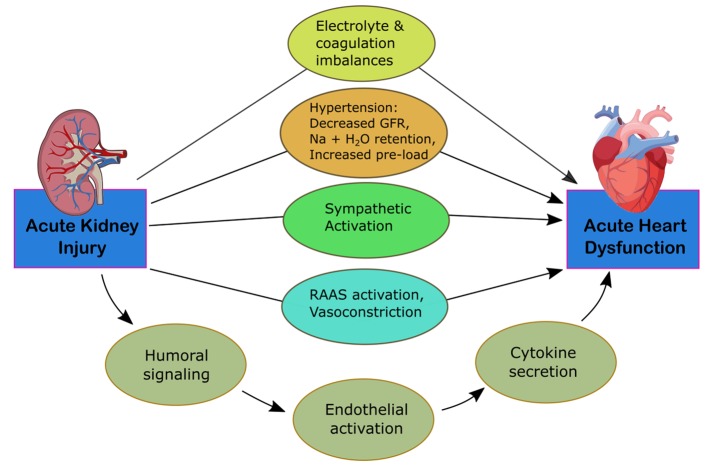
Mechanism of damage in CRS-3. CRS, cardiorenal syndrome; GFR, glomerular filtration rate; RAAS, renin-angiotensin-aldosterone system.
CRS type 4 (chronic renocardiac syndrome)
CRS-4 is characterized by the primary presence of CKD leading to cardiac disruption and is illustrated in Figure 4 [4]. Renal dysfunction is an independent risk factor for cardiovascular disease as CKD patients present a higher risk of mortality for myocardial infection and sudden death as established by Menzter et al [17]. CKD patient incident rates (per 100 patient years) were 30.7 for CHF, 3.9 for acute myocardial infarction (MI) and 35.7 for atherosclerotic vascular disease [18]. As CKD progresses, cardiac function decrease occurs due to ventricular hypertrophy and diastolic dysfunction associated with increased risk of adverse cardiovascular events [19]. CKD is considered a risk factor for sudden cardiac death due to changes to volume, arterial pressure and electrolyte disturbances increasing the risk to cardiac arrhythmias. Coronary atherosclerotic heart disease, acute coronary syndrome and left ventricular hypertrophy (LVH) are present in increased rates in CKD patients with reduction of eGFR [19]. The HEMO study found that patients with end-stage renal disease (ESRD) and advanced CKD have a 40% prevalence of ischemic heart disease and a 40% prevalence of CHF [20]. The HOPE trial demonstrated that elevated microalbuminuria increased the relative risk for cardiovascular events, mortality and CHF [21]. Increasing urine albumin is a strong indicator for mortality as a 10-fold increase of albumin to creatinine ratio increases mortality by 57% [21]. Anemia, advanced glycation end product (AGE) accumulation, oxidative stress, mineral metabolism disruption and RAAS overactivation are increasingly found in CKD patients compared to the other subtypes [13]. Hyper- and hypokalemia are seen commonly among patients with CKD which can lead to decreased cardiovascular outcomes and disorders of extracellular pH, magnesium and calcium. Hypertension, dyslipidemia, chronic inflammation, primary neuropathy and diabetes mellitus lead to cardiac disruption in CKD stage 1-2 [13]. In stages 3-4, anemia, uremic toxins, electrolyte imbalance and volume overload result in disruption of cardiac function [16]. In stage 5, soft tissue calcification and erythropoietin (EPO) resistance arise as well [13].
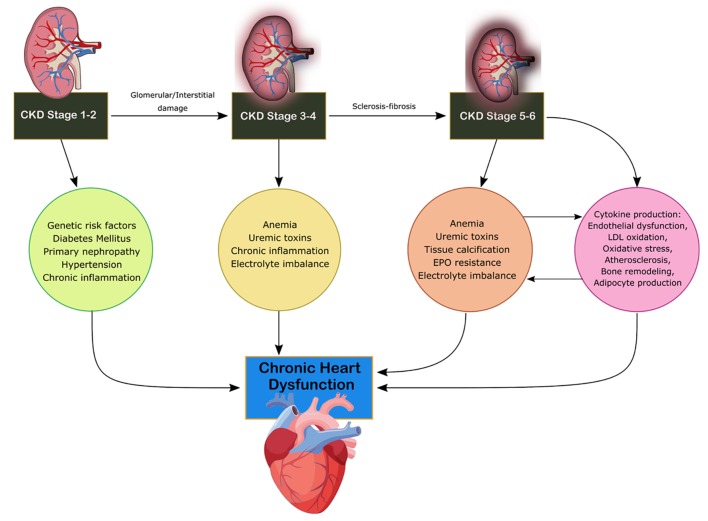
Mechanism of damage in CRS-4. CKD, chronic kidney disease; CRS, cardiorenal syndrome; EPO, erythropoietin; LDL, low-density lipoprotein.
CRS type 5 (secondary CRS)
CRS type 5 is characterized by the simultaneous presentation of cardiac and renal dysfunction as a result of an acute or chronic systemic condition and is illustrated in Figure 5. The most common condition that leads to CRS-5 is sepsis; others such as amyloid or vasculitis are less prevalent [5]. Compared to the other types of CRS, there is significantly less literature on CRS-5. The presentation of CRS type 5 varies depending on the underlying systemic condition and can be subdivided into acute and chronic conditions. Acute type-5 CRS is diagnosed as hyper-acute (0 – 72 h after diagnosis), acute (3 – 7 days), or subacute (7 – 30 days) and chronic (beyond 30 days). Acute CRS type-5 is usually superimposed above underlying cardiac and kidney disease and dependent on the systemic disease of interest [16]. This form of CRS is reversible if the systemic disease is controlled. Chronic type-5 CRS follows a variable time sequence; there is an underlying condition and causational effect that leads to the recognition. This type of CRS usually arises based on adaptive changes the body undertakes in order to combat a systemic condition. Cirrhotic patients, for example, are subject to infections and an acute type-5 CRS can overlap as a result [22]. The literature on CRS type-5 analyzes the hyper-acute phase of acute CRS type 5 as most studies evaluate the effects of sepsis [13]. AKIs can be a predicative cause of sepsis as it occurs in 20% of patients who are critically ill and in 51% of patients who experience septic shock [23]. During sepsis, renal blood flow decreases leading to acute tubular necrosis, reduction in glomerular filtration and severe kidney failure. CRS-5 is seen in 67% to 76% of sepsis patients. These patients exhibited greater severity of symptoms and an increased need for continuous renal replacement therapy and ventilator support [23].
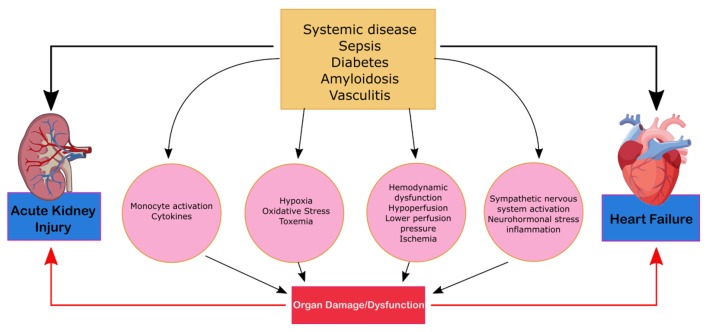
Mechanism of damage in CRS-5. CRS, cardiorenal syndrome.
Risk factors and diagnosis
Given the shared vasculature of the cardiac and renal system, dysfunction within one system becomes a risk factor of dysfunction in the other. Renal risk factors (CKD, electrolyte imbalance, elevated creatinine level, hyperkalemia and AKI) have an impact on decreasing cardiac functions. These risk factors are more indicative of CRS-1 and CRS-2 as both subtypes have primary occurrences of renal dysfunction. The outcomes study in heart failure (POSH) prospectively followed 299 patients with LV ejection factors of > 40%, baseline serum creatinine levels independently predicated WRF [24]. Examples of cardiac dysfunction (hypertension, decreased ejection fraction, acute pulmonary edema, history of HF, atrial fibrillation and atherosclerosis) lead to WRF.
The Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catherization Effectiveness (ESCAPE) trial examined 433 patients and found that the history of diabetes and hypertension was associated with an increase in SCr of 0.3 mg/dL [25]. Baseline renal insufficiency was found to be a predictor of worsening clinical outcomes. Hypertension can disrupt normal renal blood flow, which damages the vessels in the kidney and cause susceptibility to WRF [26]. The ESCAPE trial found that an improvement or a decline in eGFR during the treatment of ADHF had similar outcomes. Compared to patients with a stable GFR, those with fluctuating GFR were significantly more likely to have a reduced cardiac index, require intravenous inotrope and vasodilator therapy, and had a significantly higher rate of mortality [1]. In CRS-5, primary risk factors include sepsis, vasculitis and other systemic conditions (amyloidosis and obesity) lead to the simultaneous presentation of cardiac and renal dysfunction. Forman et al found in a retrospective study of 1,004 heart failure patients that diabetes mellitus was independently associated with higher risks of WRF [27]. Obesity is also a primary risk factor for HF and ESRD. The relative risk factor for obese individuals is 3.57 compared to 1.87 for individuals with a normal body mass index [28].
There are many renal and cardiac biomarkers that can be detected. Cardiac biomarkers are displayed in Table 2 and include B-type natriuretic peptide (NT-proBNP), inactive N-terminal pro B-type natriuretic peptide, indoxyl sulfate, cardiac troponin I (cTnI) and N-terminal propeptide of type III collagen (PIIINP). The elevated serum presence of these biomarkers indicates myocardial injury as a precursor to HF [29].
Table 2
| Biomarkers | Characteristics of presentation/site of origin |
|---|---|
| Molecular biomarkers | |
| Cardiac troponin I (cTnI) | Myocardial injury |
| B-type natriuretic peptide (BNP) | Myocardial stretching |
| sST2 | Member of interleukin (IL)-1 family of receptors |
| Indoxyl sulfate | Extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK) and nuclear factor-kappa B (NF-κB) |
| N-terminal propeptide of type III collagen (PIIINP) | Connective tissue injury |
| Physiological biomarkers | |
| Echocardiogram | Abnormal left ventricular hypertrophy |
| Central venous pressure | |
| Pericardial effusion | |
| Valvular stenosis | |
| Myocardial injury | |
| Fluid overfill | |
| Valvular calcification | |
| Doppler | Intraparenchymal blood flow that is associated |
| Ultrasound | Fluid overload |
| Chest radiograph | Cardiomegaly |
| Interstitial edema | |
| Enlarged pulmonary artery | |
| Pleural effusion | |
| Prominent superior vena cava | |
| Kerley line | |
| Kidney biomarkers | |
| Serum creatinine | Skeletal muscle |
| Albuminuria | Marker of glomerular integrity/procalcitonin (PCT) disruption |
| Kidney injury molecule (KIM-1) | Type 1 cell membrane glycoprotein expressed in regenerating PCT epithelium |
| Liver type fatty acid binding protein | Tubular injury |
| IL-18 | Cytokine mediating inflammation and AKI through the NF-κB pathway |
| Advanced glycation end products (AGE) | Improper renal clearance, myoc |
| Physiological biomarkers | |
| Ultrasound | Kidney enlargement |
| Thin and hyperechogenic cortex | |
| Small dilation of the urinary tract | |
| Parapelvic and subcortical cysts | |
| Doppler | Intraparenchymal blood flow |
Table 2 illustrates renal biomarkers of dysfunction. The most common method to detect renal injury is through the measurement of serum creatinine and albumin levels. In a prospective study by Blecker et al, those with urinary albumin to creatinine ratios (UACRs) of greater than 5 mg/g displayed progressive increases in risk for heart failure as the ratio increased. Intermediate-normal UACR (5 – 9 mg/g) was associated with a 54% increase in the relative hazard of heart failure compared to normal UACR. High-normal UACR (10 – 29 mg/g) was associated with a 91% increase in the relative hazard for heart failure [30]. Furthermore, echocardiograms, Doppler, ultrasound and chest radiograph can be used to capture images of cardiac and kidney damage. Specific areas of interest can be found in Table 2, but common markers include myocardial injury, fluid overload and kidney enlargement [1].
Treatment and management
Diuretics
While diuretics provide short term relief of patient’s symptoms, they can also cause complications such as hypertension, increased intra-abdominal pressure and renal congestion. Doses need to be properly monitored; at high doses, diuretics may enhance electrolyte imbalances, decrease circulating volume of fluid, disrupt neurohormonal balance and decrease renal function [1, 4, 13]. While diuretics are commonly prescribed (about 90% of patients with AHF), evidence-based clinical practice in HF remains uncertain with no benefit in short- or long-term mortality or re-hospitalization [1]. Loop diuretics (furosemide, bumetanide, torsemide and ethacrynic acid) represent the primary class of HF management [1]. Their effects on neurohormonal activation and renal and systemic hemodynamics can predispose to kidney injury. Decreasing kidney function in AHF results in higher rehospitalization rates and mortality. A post hoc analysis of three randomized clinical trials, DOSE-AHF (Diuretic Optimization Strategies Evaluation in Acute Heart Failure), CARRESS-HF (Cardiorenal Rescue Study in Acute Decompensated Heart Failure) and ROSE-AHF (Renal Optimization Strategies Evaluation in Acute Heart Failure) examined 198 patients who developed type 1 CRS and compared a urine volume goal-directed stepwise diuretic algorithm and standard diuretic therapy [31-33]. A stepwise algorithm directed at 24-h urine volume containing furosemide with or without metolazone resulted in an improvement in renal function compared with standard diuretic therapy [1]. The effects of low-dose dopamine, nesiritide, or placebo on decongestion and renal function were compared by ROSE-AHF [33]. No significant differences in the incidence of type 1 CRS were observed in patients’ symptoms or change in renal function. These results imply that low-dose dopamine failed to decrease congestion or to improve renal function when co-administered with diuretics [34]. The DOSE-AHF trial randomized 308 patients with AHF to bolus compared with continuous infusions of furosemide, and a low-dose compared to a high-dose regimen. High-dose diuretics (0.125 mg/day) were associated with higher rate of in-hospital WRF (65% vs. 29%) [34, 35]. It is unknown whether there is a diuretic synergy between HF and CRS. There is no evidence of a clinical trial of thiazide-type diuretics as an adjunct to furosemide in HF or CRS. The ESCAPE trial found that the use of loop diuretics did impact renal outcomes. This effect was seen irrespective of baseline kidney function, but WRF was more prevalent with the use of thiazide diuretics when the eGFR was > 60 mL/min (48% vs. 29%). This finding could tell the severity of heart or kidney failure, as thiazide is generally used when the diuretic effects of loop diuretics are insufficient. Post hoc analysis data suggest an association of escalating doses of diuretics with CRS; causation is not definitive [36].
Diuretic resistance
Diuretic resistance, a failure to achieve the therapeutically desired reduction in edema, leads to renal impairment, increased risk of hospitalization after HF and mortality. HF can prolong time to maximize concentration of drug levels [1, 37]. Loop diuretics are 95% protein bound; as such, hypoalbuminemia increases the volume of distribution and reduces the availability of loop diuretics for facilitated diffusion. Nonsteroidal anti-inflammatory drugs and uremic toxins can competitively inhibit drug through epithelial cells [1]. Repeated use of diuretics in CRS type 1 and 2 can lead to the braking phenomenon. This effect occurs when successive doses of diuretics have diminished returns. The mechanism has not been elucidated but supposes that the upregulation of distal and proximal sodium transporters causes sodium loss [38].
Ultrafiltration
Ultrafiltration is a treatment that passes blood through hollow fibers and causes the removal of isotonic fluid. The composition of ultrafiltration contrasts with the lesser sodium content in the urine produced by loop diuretics along with decongestion [39]. Without the use of loop diuretics, the potential benefits include less potassium waste, decreased renin and aldosterone release, and increased sodium loss. However, in a follow-up analysis of DOSE-AHF and CARRESS-HF, high-dose loop diuretic therapy did not result in RAAS activation greater than that with low-dose diuretic therapy. Ultrafiltration resulted in a greater increase in plasma renin activity than stepwise pharmacological care. Neither plasma renin activity nor aldosterone was significantly associated with short-term outcomes in AHF and CRS [32]. The UNLOAD, RAPID-CHF and CARRESS-HF trials all compared ultrafiltration to diuretic administration in ADHF patients. The UNLOAD and RAPID-CHF trials demonstrated that ultrafiltration was associated with a large rate of fluid loss when compared to diuretic administration, but no changes in the serum creatinine were observed. The CARRESS-HF trial evaluated the differences between ultrafiltration and pharmacologic therapy in patients who had both WRF and consistently high levels of congestion. The CARRESS-HF is the only trial that represents patients with type 1 CRS. No significant differences in weight loss were found [1]. The ultrafiltration group increased the serum creatinine of 0.23 mg/dL versus a decrease of 0.04 mg/dL in the diuretic group. Higher rates of adverse events were observed as well (72% versus 53%). Ultrafiltration therapy saw no increased benefits when compared to stepped pharmacologic therapy. Levels of weight loss remained similar between the two therapies [40, 41]. These results were confirmed by the AVOID-HF (Aquapheresis Versus Intravenous Diuretics Hospitalizations for Heart Failure) trial in 224 patients, researchers found non-significant correlation for reduced HF readmissions at 90 days, but an increase in adverse events remained (14.6% versus 5.4%) [1]. Study results and outcomes are illustrated in Supplementary Material 1 (www.cardiologyres.org).
Neurohormonal modulation and vasodilators
RAAS inhibition in chronic CRS
To counteract neurohormonal harms of CRS, RAAS inhibitors have been used to reduce mortality. The use of angiotensin-converting enzyme (ACE) and angiotensin II receptor blockers have been shown to improve survival rates for patients with HF and prevent the decrease of renal dysfunction. An improper dosage or treatment plan can lead to both increased serum creatinine levels and hyperkalemia [42]. Patients on RAAS inhibitors should begin with low dosages and have their kidney function monitored closely. RAAS studies have demonstrated beneficial effects on long-term outcomes despite an early reduction in renal function [42]. The Heart failure Endpoint evaluation of Angiotensin II Antagonist Losartan (HEAAL) trial study demonstrated that an increase in dosage from 50 mg of angiotensin receptor blocker (ARB) losartan to 150 mg retained the net clinical benefit of association with reduced risk of death or HF hospitalization despite added risk of increased serum creatinine and higher long-term reduction in eGFR [43]. The decline of GFR at initiation of RAAS may reflect antagonism of angiotensin II mediated efferent arteriolar constriction [43]. The mechanism of renal function reduction in HF patients is important in determining its prognostic significance. The Studies of Left Ventricular Dysfunction (SOLVD) investigation found early onset of decreasing renal function to be associated with increased mortality of the overall population. Within the enalapril (ACE inhibitor) group, continued use, despite an early reduction in renal function, provided a significant survival benefit. These studies suggest that decreasing renal function is not always indicative of adverse clinical outcome. In the case of ACE inhibitor administration, the manifestation of the agent’s pharmacologic properties provides a favorable effect on long-term outcomes [44]. An analysis by Ahmed et al of 1,340 patients with reduced ejection factor and eGFR of < 60 mL/min/1.73 m2 found that ACE inhibitor and ARB were associated with significant reductions in mortality [45]. The full list of studies analyzed is displayed in Supplementary Materials 2 and 3 (www.cardiologyres.org); however, a general trend seen across all studies is the general reduction of all-cause mortality amongst ACE inhibitor and ARB alone. In combination, they showed no increased clinical benefits while simultaneously displaying additional severe adverse effects (SAEs). While more prevalent in the ARB than ACE inhibitor treatment groups, renal dysfunction, hyperkalemia and hypotension were more common across both drug groups.
Vasopressin/vasodilators
Vasodilators reduce pulmonary congestion and myocardial oxygen consumption through decreasing of CVP and ventricular filling pressure [1]. A common vasodilator, intravenous nitroglycerin, treats ADHF and reduces trans-renal perfusion pressure via the reduction of venous pressure [46]. Vinod et al wrote extensively on the roles of vasopressin in HF, and found that tolvaptan can be used to treat HF in both the short and long term [47]. Furthermore secondary benefits such as weight loss, increased urine output and return to kidney function and serum electrolytes have been noted [47].
Mullens et al found that patients treated with sodium nitroprusside saw improvement in hemodynamic measurements during hospitalization along with lower rates of all-cause mortality (29% vs. 44%) and no increase in rehospitalization (58% vs. 56%) [48]. The EVEREST (Efficacy of Vasopressin Antagonism in heart Failure Outcome Study with Tolvaptan) found that the use of vasopressin tolvaptan had a positive correlation for decreasing renal function in the hospital, with return to baseline in a month. When compared to low-dose ARB administration in CHF, eGFR reductions occurred more prominently with high-dose losartan. However, high-dose losartan is associated with improved long-term health outcomes [49].
Adenosine antagonists are a secondary option crucial for patients with HF. By blocking adenosine A1 receptors, while allowing binding of A2 receptors, urine output can increase and maintain proper kidney function. The PROTECT study investigated rolofylline, an adenosine A1 receptor antagonist in AHF patients with renal dysfunction showing that neurologic adverse events were more frequent in patients receiving rolofylline [4]. No significant difference was found between cardiac or renal outputs. Renal impairment was found in 15.0% of patients in the rolofylline group and in 13.7% of patients in the placebo group. Mortality by renal or cardiac causes was similar between both the trial and the placebo group (30.7% and 31.9%) respectively. Adverse event rates were similar overall; however, only patients in the rolofylline group developed seizures [50].
Mineralocorticoid receptor antagonists (MRAs)
Suppressions of RAAS activity with ACE inhibitors and ARBs are limited by aldosterone escape, which results in increased levels of serum aldosterone. MRAs can limit this effect when added to an ACE inhibitor/ARB, and provide improved suppression of RAAS with additional cardiorenal benefits. The results from all the trials and studies analyzed are found in Supplementary Material 4 (www.cardiologyres.org). Significant outcomes noticed were the reduction in mortality and cardiovascular events with HF with a reduced ejection factor [51, 52]. The mortality rate decreased by 30% in the RALES study; however, the incidence of WRF was higher (17% vs. 7%) [52]. The EPHESUS study demonstrated a 15% reduced mortality rate with MRAs in LV dysfunction and complicating acute MI. The EMPHASIS-HF trial (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure), in which 33% of the patients had an eGFR < 60 mL/min/1.73 m2, showed that the effect of MRA eplerenone on the HF or cardiovascular death was reduced versus the placebo group (18.3% vs. 25.9) though they had higher rates of hyperkalemia (8.0% vs. 3.7% [53].
Cardiac resynchronization therapy (CRT)
CRT is a treatment that uses a pacemaker to improve ventricular contraction and reduce the risk of mitral regurgitation. A meta-analysis of 14 randomized control studies with patients diagnosed with LV systolic dysfunction showed that CRT significantly improved LV ejection, quality of life, and reduced mortality by 22% [54]. The MIRACLE study (Multicenter InSync Randomized Clinical Evaluation) evaluated CRT in HF in patients with EF < 35% and reduced renal function. While this trial excluded patients with a serum creatinine level of > 3 mg/dL, CRT increased eGFR and reduced blood urea nitrogen in individuals with eGFR of 30 ≤ eGFR< 60 compared to the control group [55]. This benefit likely resulted from improved perfusion and reduced venous congestion. Bazoukis et al showed a higher risk of mortality in patients with baseline CKD undergoing CRT. Patients with baseline eGFR < 60 mL/min/1.73 m2 had an increased mortality risk from all causes compared with patients with eGFR > 60 mL/min/1.73 m2 [56]. Supplementary Material 4 (www.cardiologyres.org) illustrates the results of trials analyzed for CRT.
Mechanical circulatory support and kidney function
While the use of mechanical circulatory support devices is prevalent in cardiogenic shock and circulatory support during high-risk coronary interventions, their effect on renal function is unknown. Flaherty et al showed a reduction in AKI rates with a percutaneous ventricular assist device during percutaneous coronary interventions [57]. The effects of continuous versus pulsatile LV assist devices on renal morphology and physiology have been described in animal models only [58]. In animals, reduced pulsatile circulation can activate local RAAS, enabling proinflammatory effects and increased vascular stiffness. Hypertrophy of renal cortical arteries, interstitial nephritis and periarteritis have been observed in animal models of continuous perfusion [53]. Welp et al demonstrated lower levels of renin and angiotensin in subjects with pulsatile- versus continuous-flow LV assist device’s plasma renin activity was substantially elevated in patients with non-pulsatile LV support. Plasma aldosterone levels were also significantly higher in patients supported by non-pulsatile LV assist devices [59]. The long-term clinical impacts have not been elucidated. Studies analyzed are shown in Supplementary Material 5 (www.cardiologyres.org).
Beta-blockers
Beta-adrenergic blockers are drugs that alleviate blood pressure via the blocking of epinephrine. Their effects have improved cardiac outlook and reduced hospitalization [1]. The MERIT-HF study (Metoprolol CR/XL Controlled Randomized Intervention Trial in Chronic HF) randomized 3,991 patients with HF and ejection factor < 40% to β-blocker metoprolol versus placebo. A secondary analysis during the study looked at beta blockade across eGFR ranges of > 60, 45 – 60 and < 45 mL/min/1.73 m2. This trial showed significant benefits across all subgroups of eGFR with those in the GFR < 45 mL/min/1.73 m2 range displaying a nearly 60% reduction in HF and mortality [60]. In the SENIORS study (Study of the Effects of Nebivolol Intervention on Outcomes and Rehospitalization in Seniors with Heart Failure), mortality risk and cardiovascular hospital admissions were significantly reduced in patients with HF who were randomized to nebivolol versus placebo [61]. Mortality or cardiovascular admission for nebivolol was 40% in < 45 mL/min/1.73 m2 group, 31% in 45 – 60 mL/min/1.73 m2 group and 29% in the > 60 mL/min/1.73 m2 group [61]. A meta-analysis of six randomized control trials with β-blockers in patients with CKD and HF showed that β-blockers significantly reduced the risk of all-cause and cardiovascular mortality. An increased risk of bradycardia and hypotension was observed with the use of beta-blockers [62]. Tolerability of β-blockers is limited by fluid retention in the heart. The MERIT-HF trial presented similar tolerance of the treatment SENIORS trials, however, found that the rates of β-blocker discontinuation were higher the lower the eGFR was [63].
Source: https://www.ncbi.nlm.nih.gov/pmc/