A Case of Ketron-Goodman Disease
M. Carlesimo
aU.O.C. Dermatology, II School of Medicine University of Rome ‘Sapienza’, Rome, Italy
A. Tammaro
aU.O.C. Dermatology, II School of Medicine University of Rome ‘Sapienza’, Rome, Italy
C. Cox
bU.O.C. Hematology, II School of Medicine University of Rome ‘Sapienza’, Rome, Italy
E. Mari
aU.O.C. Dermatology, II School of Medicine University of Rome ‘Sapienza’, Rome, Italy
L. Fidanza
aU.O.C. Dermatology, II School of Medicine University of Rome ‘Sapienza’, Rome, Italy
A. Narcisi
aU.O.C. Dermatology, II School of Medicine University of Rome ‘Sapienza’, Rome, Italy
C. Cacchi
cUnit of Histopathology, II School of Medicine University of Rome ‘Sapienza’, Rome, Italy
G. Camplone
aU.O.C. Dermatology, II School of Medicine University of Rome ‘Sapienza’, Rome, Italy
Abstract
Pagetoid reticulosis (PR) is a rare form of cutaneous T-cell lymphoma [Mod Pathol 2000;13:502–510]. Two variants of the disease are described: the localized type Woringer-Kolopp disease (WKD) and the disseminated type Ketron-Goodman disease (KGD). KGD may have disseminated lesions, high rate of recurrence and a guarded prognosis [Mod Pathol 2000;13:502–510]. In patients with KGD, therefore, long-term observation is necessary. Disappearance of cutaneous lesions does not mean resolution of the disease [J Am Acad Dermatol 2002;47:183–186]. Herein we report the case of an 84-year-old man with erythematous patches of the trunk and the upper and lower extremities in whom the diagnosis of KGD was made. We describe this case for the rarity of this pathology and for the good response to therapy (IFN).
Introduction
Pagetoid reticulosis (PR) is an uncommon disease clinically characterized by the presence of solitary, psoriasiform, slowly growing cutaneous plaques located on the extremities and, at histological level, by a characteristic dense inflammatory infiltrate with pagetoid spread within the dermis and the upper dermis composed of atypical lymphocytes. The epidermis shows a hyperkeratotic and acanthotic pattern. Two clinical variants of PR may be distinguished: a localized form, also known as Woringer-Kolopp disease (WKD), which consists of a single lesion, usually located on the extremities, presenting a good prognosis when it can be totally removed by excision or high-dose radiation, and the generalized type or Ketron-Goodman disease (KGD) with disseminated lesions and a poor prognosis [1, 2]. In the WHO and EORTC classification of cutaneous lymphomas in fact, WKD is classified as a relatively indolent variant of mycosis fungoides (MF), whereas KGD, which is not classified yet, is generally considered an aggressive lymphoma with bad prognosis similar to the aggressive CD8-positive cutaneous T-cell lymphoma, the cutaneous γ/δ-positive T-cell lymphoma and the tumor stage of MF [3].
We present a case of an 84-year-old man with a 2-year history of erythematous patches located on the trunk and the upper and lower extremities in whom the diagnosis of KGD was made.
Case Report
In April 2008 an 84-year-old man presented with a 2-year history of asymptomatic erythematous patches located on the trunk and the upper and lower extremities. On physical examination several round to oval lesions presented regular, well-defined and unindurated borders with mild desquamation and mild infiltration, associated with itching (fig. 1). He denied any dermatologic diseases and his personal history was unremarkable except for hypertension and diabetes. The patient was admitted to the Department of Dermatology of Sant’Andrea University Hospital. General examination and laboratory data were within normal limits.
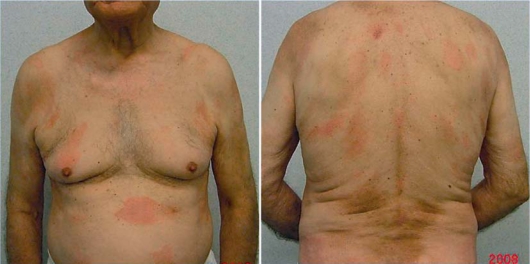
Plaque lesions with mild desquamation and induration on the trunk.
A skin biopsy (fig. 2) showed marked acanthosis of the epidermis with infiltration of atypical lymphoid cells in a pagetoid pattern. The dermis contained a band-like infiltrate of lymphoid cells displaying epidermotropism. The phenotype of neoplastic cells was CD3+ (80%), CD5+ (25%), CD45+ (85–90%), CD2 negative. Pautrier microabscesses were also visible.

a Psoriasiform hyperplasia, as seen in this picture, is a characteristic feature (hematoxilin and eosin, 10×). b Inflammatory CD45+ cells are both histiocytes and lymphocytes in the dermis (immunostaining for CD45). c T lympocytes (CD4+) located in the lower part of the epidermis (immunostaining for CD4+, 40×).
Chest X-ray, echosonography of the abdomen and bone marrow examination revealed no systemic involvement. The long clinical presentation, the typical histological examination and immunohistochemistry led to the diagnosis of KGD. The patient was treated with three cycles of INF therapy (0.5 mg/kg/day) for 2 months that led to a regression of the skin lesions with apparent complete remission until now. During 8 months of follow-up no significant changes of this clinical picture were observed.
Discussion
PR is a rare form of cutaneous T-cell lymphoma with a striking epidermotropism on histological examination [3, 4]. The term PR has been introduced by Braun-Falco et al. in 1973 to identify this clinical entity [5], first described by Woringer and Kolopp [6] in 1939, for the resemblance of infiltrating cells characterizing this condition with Paget’s cells present in the epidermotropic infiltrate of mammary Paget’s disease. Different authors proposed various etiopathogenetic hypotheses for this disease: at the beginning it was considered as a reactive lymphoproliferative process induced by different stimuli, including arthropod bite and with evolution towards neoplastic disease [7]. Other authors considered PR as a variant of cutaneous T-cell lymphomas [8, 9, 10]. The origin of the pagetoid cells has been uncertain for many years: melanocytes, histiocytes, Merkel cells, true Paget cells and Langerhans cells were all proposed, but with the advent of phenotypical studies a T-cell lineage was finally confirmed [11].
The classic skin lesions usually begin with asymptomatic scaly round or oval plaques that can be erythematous or yellowish with regular, well-defined and unindurated borders, associated with desquamation. Lesions typically affect the trunk and the extremities. The basic criterion of diagnosis is the presence of an inflammatory infiltrate composed of atypical lymphoid cells within the epidermis and the upper dermis with pagetoid distribution. The histologic features of KGD are identical to those of WKD. The difference between the two variants is based on clinical grounds alone [4].
The relationship between PR and MF is still unclear; in fact different authors consider the first as a clinical variant of the second. Haghighi et al. in 2005 [12] suggested that PR might be classified as a cutaneous T-cell lymphoma with uncertain clinical behavior, considering it as a distinct clinicopathologic entity, separate from MF. In fact there are several clinical, histologic and immunophenotypic differences between these two entities. A more difficult distinction is that of PR by mycosis palmaris et plantaris, a rare variant of MF, characterized by acral plaques resembling those of WKD. Therefore there are several features that make PR unique among the classification of cutaneous T-cell lymphomas: the acral distribution of lesions, the pronounced hyperkeratosis and acanthosis at histology, the prominent epidermotropism and the expression of αEβ7 integrin on T-lymphocytes that binds cadherins on keratinocytes [4]. Some authors have proposed that epidermotropism of lymphoid cells is probably due to the expression of integrin αEβ7 on the cell surface, which interacts with E-cadherin on epithelial cells [2]. In fact expression of αEβ7 on lymphocytes is often increased in those inflammatory conditions in which T-cells infiltrate epithelial tissues. The function of αEβ7 is not yet fully understood, but it is likely to be important in the retention of T-cells in mucosal tissues and may also have a role in cell signaling and heterotypic communication between lymphocytes and epithelial cells [13].
In PR three prevalent phenotypes of neoplastic cells were described: CD4+ (T-helper phenotype), CD8+ (T-cytotoxic/suppressor phenotypes) or CD4/CD8 double negative phenotype. Some cells could be CD30+ and do not express CD45RO [4]. Neoplastic cells can express other CD45 isoforms (CD45RA) in the disseminated aggressive variant (KGD) but the complete CD45 family is lost from the cell surface in the benign localized variant (WKD). Considering the fact that the presence of CD45 is crucial for activation of the lymphocyte-specific tyrosine kinase (p561ck), which drives lymphocyte growth and transformation, lack of this molecule may be the molecular basis for the non-aggressive nature of this clonal T-cell proliferation [11].
In our case histological and phenotypical analysis suggested a diagnosis of KGD and, in particular, the presence of rare CD30+ cells and a component of Langerhans cell type within the epidermis with a CD1a+ phenotype (30%) has also been reported [12]. So, clinical picture, histologic findings and molecular studies confirmed the diagnosis of KGD.
Only a few cases of KGD are described in the literature; therefore we report this case for the rarity and for the unexpected complete regression of the skin lesions after IFN therapy. Although KGD is characterized by an high rate of recurrence, during 8 months of follow-up our patient is in good condition without evidence of recurrent lesions. At this time we cannot exclude the possibility of recurrences and long-term observation is necessary.
References
Content retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2895208/.