Heart failure after myocardial infarction in the era of primary percutaneous coronary intervention: Mechanisms, incidence and identification of patients at risk
Thomas J Cahill and Rajesh K Kharbanda
World J Cardiol. 2017 May 26; 9(5): 407–415. Published online 2017 May 26. doi: 10.4330/wjc.v9.i5.407 PMCID: PMC5442408PMID: 28603587
Received 2016 Oct 30; Revised 2017 Jan 20; Accepted 2017 Mar 12.
Original source
Abstract
Myocardial infarction (MI) remains the most common cause of heart failure (HF) worldwide. For almost 50 years HF has been recognised as a determinant of adverse prognosis after MI, but efforts to promote myocardial repair have failed to translate into clinical therapies. Primary percutaneous coronary intervention (PPCI) has driven improved early survival after MI, but its impact on the incidence of downstream HF is debated. The effects of PPCI are confounded by the changing epidemiology of MI and HF, with an ageing patient demographic, an increasing proportion of non-ST-elevation myocardial infarction, and the recognition of HF with preserved ejection fraction. Herein we review the mechanisms of HF after MI and discuss contemporary data on its incidence and outcomes. We review current and emerging strategies for early detection of patients at risk of HF after MI, with a view to identification of patient cohorts for novel therapeutic agents.Keywords: Angioplasty, Heart failure, Myocardial infarction, Percutaneous coronary intervention, ST-elevation myocardial infarction
Core tip: Heart failure (HF) is a major cause of late morbidity and mortality after myocardial infarction. Several approaches exist for early identification of patients at risk of HF, including clinical and angiographic scoring, cardiac imaging, and invasive coronary physiology, but these are currently poorly integrated. Here we provide an overview of the incidence, mechanisms, and outcomes of HF following myocardial infarction in the era of primary percutaneous coronary intervention, and discuss HF risk-stratification for the individual patient. Looking ahead, accurate and early prediction of HF will allow targeting of novel therapeutic agents to high-risk patients before ventricular remodelling and clinical HF have become established.Go to:
INTRODUCTION
Primary percutaneous coronary intervention (PPCI) has revolutionised the management and outcome of acute myocardial infarction (MI)[1,2]. It is the reperfusion strategy of choice throughout the developed world, with 90000 procedures performed annually in the United States alone[3,4]. Contemporary PPCI in the United Kingdom is characterised by door-to-balloon times of < 60 min, radial access, second generation drug-eluting stents and tailored use of antiplatelet and antithrombotic agents[5,6]. The introduction of PPCI and adjunctive therapies have driven a reduction in inpatient mortality following acute MI from 20% in the late 1980s to approximately 5%-7% in contemporary series[7,8].
Despite this success, coronary artery disease remains the commonest cause of heart failure (HF)[9]. HF after MI is the major driver of late morbidity, mortality and healthcare cost. The effect of PPCI on the incidence of HF is debated, with studies confounded by the changing definitions and epidemiology of both MI and HF. Targeted therapies to prevent HF after MI have lagged behind advances in reperfusion, with Entresto (valsartan/sacubitril) the only novel pharmacotherapy for HF to enter the mainstream market in over a decade. Despite the evident burden of HF, many MI trials have predominantly focused on thrombosis, bleeding and composite endpoints [e.g., major adverse cardiac events (MACE)] rather than HF events specifically.
In this review we outline the challenge of HF following MI in contemporary practice. We provide an overview of the mechanisms and definitions of HF after MI, and the data on temporal trends in HF incidence from the pre-thrombolysis era through to the modern day. We review current and emerging strategies to identify patients at risk of HF, including coronary physiology, cardiac magnetic resonance and hybrid imaging, and suggest how improved mechanistic understanding of HF can be used to inform the next generation of clinical trials for these patients.Go to:
DEFINING HF AFTER MI
HF is defined as “a clinical syndrome resulting from any structural or functional cardiac disorder that impairs the ability of the ventricle to fill or eject blood”[10]. This has been translated into several validated diagnostic criteria (e.g., the Framingham criteria[11] and the European Society of Cardiology criteria[12]), but the primary definition of HF as a clinical syndrome has led to differing clinical, imaging and biomarker definitions coexisting in clinical practice and research. Unlike MI, there is no “universal definition” and consequently, the diagnosis of HF is often heterogeneous between studies and over time[13].
Early studies of HF following MI used clinical criteria such as the Killip and New York Heart Association classifications[14,15]. While crude, Killip class has retained prognostic value in more recent cohorts such as the GRACE registry: Patients in Killip class I had an in-hospital mortality of 3%, rising to 20% for those in class III[16]. In the PPCI population, higher Killip class at presentation is an independent predictor of in-hospital and 6-mo mortality[17]. Clinical HF scores were refined by the development of echocardiography, which led to objective measurement of ejection fraction and ventricular volumes as an intrinsic part of a HF diagnosis[18].
The timing of HF following MI is important clinically, mechanistically and for research. Three key time periods need to be distinguished: HF at the index MI presentation, during the course of the first admission, and after discharge. The timing of HF is often not well-defined by research studies, making comparison between studies challenging. From a statistical perspective, older studies failed to treat HF as a time-dependent, evolving covariate whose incidence was modifiable by the up-front treatment, meaning that late-onset HF after MI is less well characterised than HF at presentation[19].Go to:
PATHOPHYSIOLOGY OF HF AFTER MI
Several overlapping mechanisms contribute to HF after MI (Figure (Figure1).1). HF during the index MI occurs due to a combination of myocardial stunning, myocyte necrosis, decompensation of pre-existing HF or acute mitral regurgitation due to papillary muscle dysfunction. HF during the hospitalisation may also be due to any of the above, compounded by fluid or contrast overload, renal dysfunction, or complications such as ventricular septal defect or cardiac tamponade. Late HF reflects the consequences of cardiomyocyte death and scar formation occurring alongside ventricular remodelling.
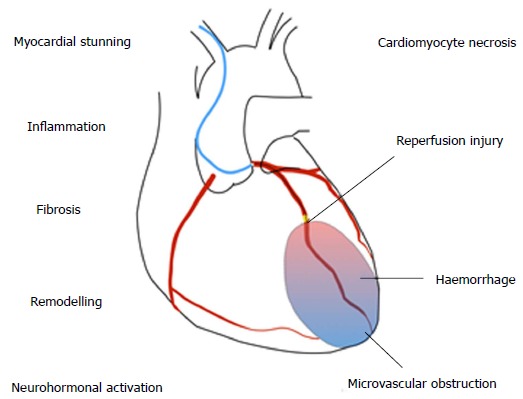
Mechanisms of heart failure after myocardial infarction.
The cellular pathophysiology of MI has been clearly defined in animal models. Within 30 min of ischaemia, cardiomyocyte structural changes and oedema develop, leading to progressive cell death from three hours. Acute contractile dysfunction occurs due to oxidative stress and calcium overload, which is reversible if flow is restored[20]. Reperfusion itself causes a second wave of injury, by production of reactive oxygen species. Despite successful epicardial reperfusion, embolization of thrombotic debris, plugging by inflammatory cells and release of vasoactive mediators from damaged endothelium leads to ongoing microvascular dysfunction in up to 50% of patients[21].
Myocardial injury leads to activation of a stereotyped inflammatory cascade, comprised of early neutrophil ingress followed by monocyte-macrophage infiltration. Between days 3-5 following MI there is a transition from inflammation to repair, with activation of fibroblasts and progressive scar deposition[22]. Over time there is compensatory activation of the renin-angiotensin and sympathetic nervous systems and pathological remodelling, with changes to the ventricular geometry, wall thinning, ischaemic mitral regurgitation and further cardiomyocyte loss. The precise contribution of the different pathophysiological components (e.g., microvascular dysfunction, inflammation) to injury is likely to be heterogeneous, and understanding mechanistic pathways in specific patient subgroups will be key to identifying novel therapeutic strategies[23].Go to:
TEMPORAL TRENDS IN HF AFTER MI
HF after MI was first described as an adverse prognostic feature by Killip in the 1960s[14]. HF was associated with large infarcts and multivessel disease, and the presence of impaired ventricular function was linked to worsening mortality[24,25]. Prior to thrombolysis, the incidence of in-hospital HF after ST-elevation myocardial infarction (STEMI) was approximately 40%[15]. This appeared to reduce after the introduction of thrombolysis, with HF present in approximately 3% of patients at presentation and 17% during admission[26]. Successful reperfusion was associated with improved LV function and long-term survival[27]. HF during admission remained an adverse prognostic feature, with 1-year mortality rates approximately 5 fold higher in those with HF[28].
More recent studies have suggested a further reduction in HF rates with use of primary PCI. In an Italian cohort of 2089 MI patients treated exclusively by PPCI between 1995 and 2005, 17% presented in HF, but only a further 1% developed new onset HF during the hospital admission[29]. Similarly, in an analysis from the HORIZONS-AMI cohort of 3602 patients recruited between 2005-2007 treated with PPCI, 8.0% of patients were in Killip class II-IV at presentation. At 30 d, only 4.6% of patients had developed a clinical HF syndrome (defined by NYHA/Killip class), rising to 5.1% at 2 years[30].
These studies are not directly comparable, and reflect selected trial cohorts with a short duration of follow-up and differing methods of HF ascertainment. Several dedicated time trend analyses have now been performed. In Olmsted County, 1537 patients with an index MI between 1979 and 1994 were identified, spanning the introduction of thrombolysis in the late 1980s[31]. Over the study period the 5-year incidence of HF decreased from 40% to 33%. In a later study of 2596 MI patients between 1990 and 2010, there was increasing use of PPCI and a reduced risk of both early (0-7 d; HR = 0.67, 95%CI: 0.54-0.85) and late (8 d-5 years; HR = 0.63, 95%CI: 0.45-0.88) HF over time[32]. In patients with HF, mortality was higher for those with delayed vs early onset HF[33].
A reduction in post-MI HF has also been seen in other studies. In a sample of 2.8 million MI hospitalisations (in Medicare beneficiaries) between 1998-2010, there was a reduction in the incidence of subsequent HF hospitalisation from 16 to 14 per 100 person-years[34]. In a Danish cohort, the incidence of HF at 90 d post-MI reduced from 24% to 20% alongside a an increase in PCI from 2.5% to 38% between 1997 and 2010[35]. Similarly, in Western Australia there was a reduction in the prevalence of HF at 90 d from 28% to 17% between 1996 and 2007[36]. Finally, in the SWEDEHEART registry of 199851 patients admitted with an MI between 1996 and 2008, there was a reduction in the incidence of clinical HF (albeit measured during the index hospitalization) from 46% to 28% alongside increasing use of PPCI[37].
In contrast, data from Framingham Heart Study shows an increase in HF over time[38,39]. In 676 patients who developed a first MI between 1970 and 1999, both the 30-d and 5-year incidence of HF increased. At 5 years, the incidence of HF was 28% in 1970-1979 and 32% in 1990-1999, which remained significant after multivariate adjustment, with a risk ratio of 1.74 (95%CI: 1.07-2.84) for HF in 1990-1999 compared with 1970-1979[39]. Similarly, the Worcester Heart Attack Study showed an increasing incidence of HF between 1975 and 2001, and in the Alberta Elderly MI cohort, there was a 25% increase in the incidence of in-hospital HF between 1994-2000, from 31% to 39%[40,41].
In the face of conflicting data from a relatively small number of studies, it remains difficult to draw firm conclusions on the impact of PPCI on the incidence of HF. There are key differences between studies in the definition of HF and MI, the validation of these diagnoses, the timing of ascertainment and the duration of follow-up. While the majority appear to suggest that PPCI is associated with reduced HF incidence, for many the duration of follow-up is short. Some studies have not differentiated between incident and prevalent cases. Reconciling these differences will require further studies with long term follow-up. If increasing survival from MI is leading to a rise in HF, this may only be seen in updated analyses, as interventionalists take on increasingly frail and elderly patients who previously were not surviving[42].Go to:
RISK STRATIFICATION FOR HF AFTER MI
Given the ongoing contribution of HF to morbidity and mortality after MI, early risk stratification and preventative therapeutic strategies are required. Although a number of clinical, angiographic, physiological, imaging and biomarker approaches to HF risk stratification following MI have been proposed (Figure (Figure2),2), few are in routine clinical use. In addition to prognostication, emerging hybrid imaging and coronary physiology approaches are shedding light on the mechanisms driving HF in different patient subgroups, and might also be used as early surrogate endpoints for trials. Given the historical failure of generic approaches targeting processes such as inflammation, it seems likely that tailored therapies for specific patient groups will be required.
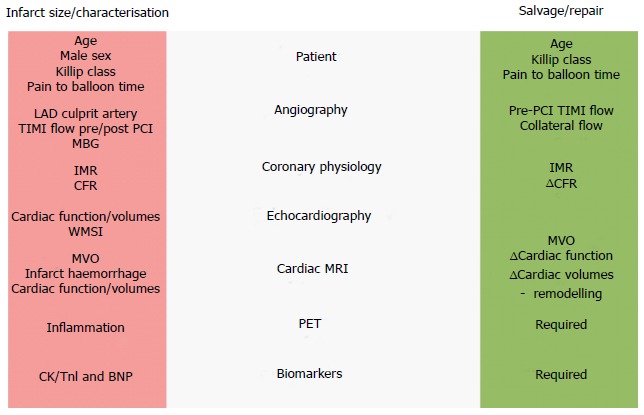
Heart failure after myocardial infarction – strategies for prediction of infarct size and salvage. BNP: B-type natriuretic peptide; CFR: Coronary flow reserve; CK: Creatine kinease; IMR: Index of microcirculatory resistance; LAD: Left anterior descending; MBG: Myocardial blush grade; MVO: Microvascular occlusion; PET: Positron emission tomography; PCI: Percutaneous coronary intervention; TIMI flow: Thrombolysis In Myocardial Infarction flow score; TnI: Troponin I; WMSI: Wall motion score index.
Clinical evaluation
Infarct size is the major determinant of downstream HF and prognosis[43]. Predictors of HF on admission are related to underlying LV function (e.g., prior MI), markers of coronary artery disease severity (e.g., diabetes mellitus) and features of an extensive infarct (e.g., anterior location, increasing duration from symptom onset to reperfusion)[29]. Clinical predictors of late HF are of greater use for risk stratification. In the HORIZONS-AMI cohort, predictors of new-onset HF at 2 years were a history of MI (OR = 1.81, 95%CI: 1.22-2.67), ejection fraction (per 10% decrease OR = 1.35, 95%CI: 1.21-1.5), female sex (OR = 1.34, 95%CI: 1.1-1.51) and insulin-treated diabetes (OR = 1.68, 95%CI: 0.96-2.65)[30]. Similarly, in the CARE and VALIANT studies of MI survivors, predictors of late HF were age, diabetes, renal insufficiency, LVEF post-MI, and Killip class at index MI ≥ 2[44,45]. A dedicated scoring system for prediction of late HF which integrated these parameters would be of value. Although not designed for HF events, the GRACE score, originally developed for ACS risk stratification, has recently been shown to predict late HF events after both STEMI and NSTEMI[46].
Coronary angiography and haemodynamics
Coronary angiography provides a number of patient-specific markers for risk stratification, albeit often for MACE rather than HF specifically (Figure (Figure2).2). The presence of multivessel disease and lack of normal flow in the infarct related artery are key adverse prognostic features after PPCI[47,48]. TIMI flow in the infarct related artery pre and post-procedure predicts outcome, with post-procedural ≤ TIMI 2 flow associated with a HR of 3.8 (95%CI: 2.5-5.7) for 1-year mortality[49]. Impaired microvascular perfusion, assessed angiographically using the TIMI myocardial blush grade, also predicts 1-year mortality, which rises from 1.4% in patients with normal blush to 6.2% in patients with absent blush[50]. Patients with no-reflow have a higher incidence of arrhythmia, remodelling, HF and mortality[21].
Recent attempts to categorise no-reflow have focused on the upstream causes, including distal embolisation of thrombus, new thrombus formation mid-procedure, and intraprocedural stent thrombosis, collectively badged as intraprocedural thrombotic events (IPTE). The incidence of IPTE was 12.2% in a recent STEMI cohort, with each IPTE component independently associated with 30-d death and MACE[51]. Beyond angiography, invasive hemodynamic measurement in the cath lab can provide further risk stratification: Measurement of LV end-diastolic pressure is an independent predictor of mortality at 2 years, even after adjustment for baseline LV function[52].
Coronary physiology
Invasive coronary physiology provides a highly sensitive readout of microcirculatory function, and has demonstrated significant value in prediction of myocardial recovery after MI. The index of microcirculatory resistance (IMR) is the ratio of distal coronary pressure to the inverse of the mean transit time during maximal hyperaemia. Measured after PPCI, IMR is a predictor of ejection fraction and infarct volume at 3 mo post-MI[53]. An IMR > 40 is associated with increased risk of death or rehospitalisation for HF (HR = 2.1, P = 0.034)[54]. IMR is now being used as a surrogate endpoint in ongoing trials of aspiration thrombectomy and intracoronary GpIIb/IIIa agents[55]. Hyperaemic microvascular resistance (HMR), another specific read-out of microcirculatory function, is also predictive of CMR-defined microvascular occlusion (MVO), and impaired local blood flow as measured by PET[56].
Invasive measurement of coronary flow reserve (CFR), zero-flow pressure (Pzf), and fractional flow reserve (FFR) may also have a role in risk stratification following PPCI. CFR is the ratio of ratio of hyperaemic to resting coronary flow and incorporates both the epicardial and microvascular circulations. In 44 patients undergoing PPCI, the change in CFR between presentation and day 1 post-PPCI was predictive of the degree of myocardial salvage and ejection fraction at 6 mo[57]. Pzf, derived from pressure-velocity loop analysis, is the distal coronary pressure at which the flow in a coronary artery would theoretically cease and represents extravascular compression of the microcirculation by oedema or haemorrhage. Pzf correlates with HMR, and also predicts residual scar at 6 mo post-MI with an AUC of 0.94[56,58]. FFR, the ratio of myocardial blood flow at maximal hyperaemia in comparison to normal proximal myocardial flow, is also predictive of adverse outcome, with an FFR of ≤ 0.8 associated with an HR of 3.24 for MACE[58]. The utility of coronary physiology in day-to-day practice, and the optimal index for HF risk stratification remains to established.
Imaging
Complimentary imaging modalities are providing increasingly detailed phenotyping of myocardial injury, function and healing. Standard echocardiographic indices including ejection fraction, LV volumes, wall motion score index, E/E’ ratio and right ventricular function provide prognostic information after MI[59]. More recently, longitudinal and circumferential strain rate have been shown to be predictive of death or hospital admission for HF, with longitudinal strain rate adding significant incremental value in the prediction of all-cause mortality beyond clinical variables, LVEF, and wall motion score index[60].
Cardiac MRI offers a broad armamentarium for tissue and functional characterisation, including quantification of the area at risk, infarct size, salvage index, microvascular obstruction, haemorrhage, heterogeneity and scar. Several indices are independently predictive of late outcome, including CMR-determined infarct size, myocardial salvage index and extent of microvascular obstruction[61–63]. In a study of 249 patients, CMR measurement of MVO was the strongest predictor of MACE over a 6-year follow-up[64,65]. CMR characterisation of infarct core characteristics, including identification of infarct haemorrhage (by T2W and T2*) or native T1 signal, are recently-described predictors of adverse remodelling and clinical outcome[66–68].
Combining metabolic imaging by 18F-fludeoxyglucose (18F-FDG) with MRI has the potential to provide further characterisation of myocardial injury and repair. 18F-FDG accumulates in monocyte-macrophages and other highly metabolically active tissues[69]. In a study of 49 patients hybrid 18F-FDG PET-MRI was performed at a median 5 d following MI. The intensity of 18F-FDG signal correlated with infarct size and predicted cardiac function at 6-9 mo follow-up. Interestingly, FDG signal remained an independent predictor after multivariate analysis, providing the first clue that imaging of inflammatory response can be used to risk stratify patients[70]. Recently, PET imaging of CXCR4, a receptor expressed on leucocytes and haematopoietic stem cells was demonstrated in human patients after MI. This type of approach holds great potential for understanding the biological heterogeneity in the healing response in vivo[71].
Biomarkers
Cardiac biomarkers such as troponin and BNP have established core use for the diagnosis of MI and HF, and also demonstrate prognostic value for long-term outcome[72]. A recent study shows that combining serial measurement of traditional biomarkers (e.g., NT-proBNP, hs-cTnT, aspartate transferase, alanine transaminase, lactate dehydrogenase and high-sensitivity C-reactive protein) gives an area under the curve of 0.85 for prediction of LV remodelling[73]. Looking forward, the field needs biomarkers which define specific biological groups at risk of HF who can be targeted with novel therapeutic agents. These might include biomarkers of inflammation, persistent fibrosis or matrix remodelling. There are data which highlight the potential value of measuring the inflammatory cascade for risk stratification: For example, the ratio of neutrophil:lymphocyte count predicts mortality after NSTEMI and STEMI, which may reflect the functional transition from inflammation to repair[74,75].
There are a number of emerging potential biomarkers for future HF, including tenascin-C, myeloperoxidase, cytokines, matrix metalloproteinases and growth factors[76]. Tenascin-C is an extracellular matrix glycoprotein which is not normally expressed in the heart, but is upregulated following MI, or in myocarditis. In patients with acute MI, peak tenascin-C level measured at day 5 is independently predictive of LV remodelling, HF and MACE, and provides additive value to TIMI score and BNP[77,78]. Copeptin, the C-terminal portion of provasopressin, was the strongest marker of HF after MI from the OPTIMAAL study: A doubling of copeptin was related to a 1.83 fold (1.26-2.64) increased risk of mortality (P < 0.0001)[79,80]. Galectin-3 has shown some promise as a marker of matrix and fibrosis, but has not been found to be an independent predictor of LV remodelling[81].Go to:
CONCLUSION
HF remains a major challenge after MI. Despite the difficulties of interpreting incidence over time, HF indisputably drives much of the late mortality after successful revascularisation and therapeutic interventions to prevent HF in patients at high risk would be hugely valuable. Trials would be facilitated by consensus definitions for HF events and imaging endpoints (e.g., oedema, myocardial salvage and remodelling). Given the long follow-up required for HF events, use of surrogate markers of HF risk (e.g., IMR) could be used for early translational studies. To date, despite an attractive window of opportunity to target myocardial healing, therapies in this field have failed to translate. In part this may reflect divergent mechanisms of HF in different patients, with varying contributions from microcirculatory dysfunction, inflammation, haemorrhage, oedema and remodelling. Coronary physiology and CMR imaging can be used to identify discrete subsets of patients, for example those with MVO, for targeted therapies. For risk stratification, the optimal combination of predictive modalities needs to be defined. Looking ahead, there are three key questions: What is the risk of downstream HF, what mechanisms can be identified, and what therapies can we trial?
Footnotes
Footnotes
Conflict-of-interest statement: We have read and understood BPG’s revision policy on declaration of interests and declare that we have no competing interests.
Manuscript source: Invited manuscript
Specialty type: Cardiac and cardiovascular systems
Country of origin: United Kingdom
Peer-review report classification
Grade A (Excellent): A, A, A
Grade B (Very good): 0
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: November 3, 2016
First decision: January 14, 2017
Article in press: March 13, 2017
P- Reviewer: Komócsi A, Sato A, Santulli G S- Editor: Song XX L- Editor: A E- Editor: Wu HL
References
- Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med. 2012;366:54–63. [PubMed] [Google Scholar]
- Smilowitz NR, Feit F. The History of Primary Angioplasty and Stenting for Acute Myocardial Infarction. Curr Cardiol Rep. 2016;18:5. [PubMed] [Google Scholar]
- Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet. 2003;361:13–20. [PubMed] [Google Scholar]
- Dehmer GJ, Weaver D, Roe MT, Milford-Beland S, Fitzgerald S, Hermann A, Messenger J, Moussa I, Garratt K, Rumsfeld J, et al. A contemporary view of diagnostic cardiac catheterization and percutaneous coronary intervention in the United States: a report from the CathPCI Registry of the National Cardiovascular Data Registry, 2010 through June 2011. J Am Coll Cardiol. 2012;60:2017–2031. [PubMed] [Google Scholar]
- Banning AP, Baumbach A, Blackman D, Curzen N, Devadathan S, Fraser D, Ludman P, Norell M, Muir D, Nolan J, et al. Percutaneous coronary intervention in the UK: recommendations for good practice 2015. Heart. 2015;101 Suppl 3:1–13. [PMC free article] [PubMed] [Google Scholar]
- Mamas MA, Ratib K, Routledge H, Fath-Ordoubadi F, Neyses L, Louvard Y, Fraser DG, Nolan J. Influence of access site selection on PCI-related adverse events in patients with STEMI: meta-analysis of randomised controlled trials. Heart. 2012;98:303–311. [PubMed] [Google Scholar]
- Jernberg T, Johanson P, Held C, Svennblad B, Lindbäck J, Wallentin L. Association between adoption of evidence-based treatment and survival for patients with ST-elevation myocardial infarction. JAMA. 2011;305:1677–1684. [PubMed] [Google Scholar]
- Puymirat E, Simon T, Steg PG, Schiele F, Guéret P, Blanchard D, Khalife K, Goldstein P, Cattan S, Vaur L, et al. Association of changes in clinical characteristics and management with improvement in survival among patients with ST-elevation myocardial infarction. JAMA. 2012;308:998–1006. [PubMed] [Google Scholar]
- Roger VL. Epidemiology of heart failure. Circ Res. 2013;113:646–659. [PMC free article] [PubMed] [Google Scholar]
- Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62:e147–e239. [PubMed] [Google Scholar]
- McKee PA, Castelli WP, McNamara PM, Kannel WB. The natural history of congestive heart failure: the Framingham study. N Engl J Med. 1971;285:1441–1446. [PubMed] [Google Scholar]
- Swedberg K, Cleland J, Dargie H, Drexler H, Follath F, Komajda M, Tavazzi L, Smiseth OA, Gavazzi A, Haverich A, et al. Guidelines for the diagnosis and treatment of chronic heart failure: executive summary (update 2005): The Task Force for the Diagnosis and Treatment of Chronic Heart Failure of the European Society of Cardiology. Eur Heart J. 2005;26:1115–1140. [PubMed] [Google Scholar]
- Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD, Katus HA, Lindahl B, Morrow DA, Clemmensen PM, et al. Third universal definition of myocardial infarction. Circulation. 2012;126:2020–2035. [PubMed] [Google Scholar]
- Killip T, Kimball JT. Treatment of myocardial infarction in a coronary care unit. A two year experience with 250 patients. Am J Cardiol. 1967;20:457–464. [PubMed] [Google Scholar]
- Spencer FA, Meyer TE, Goldberg RJ, Yarzebski J, Hatton M, Lessard D, Gore JM. Twenty year trends (1975-1995) in the incidence, in-hospital and long-term death rates associated with heart failure complicating acute myocardial infarction: a community-wide perspective. J Am Coll Cardiol. 1999;34:1378–1387. [PubMed] [Google Scholar]
- Steg PG, Dabbous OH, Feldman LJ, Cohen-Solal A, Aumont MC, López-Sendón J, Budaj A, Goldberg RJ, Klein W, Anderson FA. Determinants and prognostic impact of heart failure complicating acute coronary syndromes: observations from the Global Registry of Acute Coronary Events (GRACE) Circulation. 2004;109:494–499. [PubMed] [Google Scholar]
- DeGeare VS, Boura JA, Grines LL, O’Neill WW, Grines CL. Predictive value of the Killip classification in patients undergoing primary percutaneous coronary intervention for acute myocardial infarction. Am J Cardiol. 2001;87:1035–1038. [PubMed] [Google Scholar]
- Nicod P, Gilpin E, Dittrich H, Chappuis F, Ahnve S, Engler R, Henning H, Ross J. Influence on prognosis and morbidity of left ventricular ejection fraction with and without signs of left ventricular failure after acute myocardial infarction. Am J Cardiol. 1988;61:1165–1171. [PubMed] [Google Scholar]
- Eapen ZJ, Tang WH, Felker GM, Hernandez AF, Mahaffey KW, Lincoff AM, Roe MT. Defining heart failure end points in ST-segment elevation myocardial infarction trials: integrating past experiences to chart a path forward. Circ Cardiovasc Qual Outcomes. 2012;5:594–600. [PubMed] [Google Scholar]
- Tennant R, Wiggers CJ. The effect of coronary occlusion on myocardial contraction. Am Heart J. 1935;112:351–361. [Google Scholar]
- Niccoli G, Burzotta F, Galiuto L, Crea F. Myocardial no-reflow in humans. J Am Coll Cardiol. 2009;54:281–292. [PubMed] [Google Scholar]
- Braunwald E. Heart failure. JACC Heart Fail. 2013;1:1–20. [PubMed] [Google Scholar]
- Heusch G, Gersh BJ. The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: a continual challenge. Eur Heart J. 2017;38:774–784. [PubMed] [Google Scholar]
- Sanz G, Castañer A, Betriu A, Magriña J, Roig E, Coll S, Paré JC, Navarro-López F. Determinants of prognosis in survivors of myocardial infarction: a prospective clinical angiographic study. N Engl J Med. 1982;306:1065–1070. [PubMed] [Google Scholar]
- Risk stratification and survival after myocardial infarction. N Engl J Med. 1983;309:331–336. [PubMed] [Google Scholar]
- Hasdai D, Topol EJ, Kilaru R, Battler A, Harrington RA, Vahanian A, Ohman EM, Granger CB, Van de Werf F, Simoons ML, et al. Frequency, patient characteristics, and outcomes of mild-to-moderate heart failure complicating ST-segment elevation acute myocardial infarction: lessons from 4 international fibrinolytic therapy trials. Am Heart J. 2003;145:73–79. [PubMed] [Google Scholar]
- Sheehan FH, Doerr R, Schmidt WG, Bolson EL, Uebis R, von Essen R, Effert S, Dodge HT. Early recovery of left ventricular function after thrombolytic therapy for acute myocardial infarction: an important determinant of survival. J Am Coll Cardiol. 1988;12:289–300. [PubMed] [Google Scholar]
- O’Connor CM, Hathaway WR, Bates ER, Leimberger JD, Sigmon KN, Kereiakes DJ, George BS, Samaha JK, Abbottsmith CW, Candela RJ, et al. Clinical characteristics and long-term outcome of patients in whom congestive heart failure develops after thrombolytic therapy for acute myocardial infarction: development of a predictive model. Am Heart J. 1997;133:663–673. [PubMed] [Google Scholar]
- Santoro GM, Carrabba N, Migliorini A, Parodi G, Valenti R. Acute heart failure in patients with acute myocardial infarction treated with primary percutaneous coronary intervention. Eur J Heart Fail. 2008;10:780–785. [PubMed] [Google Scholar]
- Kelly DJ, Gershlick T, Witzenbichler B, Guagliumi G, Fahy M, Dangas G, Mehran R, Stone GW. Incidence and predictors of heart failure following percutaneous coronary intervention in ST-segment elevation myocardial infarction: the HORIZONS-AMI trial. Am Heart J. 2011;162:663–670. [PubMed] [Google Scholar]
- Hellermann JP, Goraya TY, Jacobsen SJ, Weston SA, Reeder GS, Gersh BJ, Redfield MM, Rodeheffer RJ, Yawn BP, Roger VL. Incidence of heart failure after myocardial infarction: is it changing over time? Am J Epidemiol. 2003;157:1101–1107. [PubMed] [Google Scholar]
- Gerber Y, Weston SA, Berardi C, McNallan SM, Jiang R, Redfield MM, Roger VL. Contemporary trends in heart failure with reduced and preserved ejection fraction after myocardial infarction: a community study. Am J Epidemiol. 2013;178:1272–1280. [PMC free article] [PubMed] [Google Scholar]
- Gerber Y, Weston SA, Enriquez-Sarano M, Berardi C, Chamberlain AM, Manemann SM, Jiang R, Dunlay SM, Roger VL. Mortality Associated With Heart Failure After Myocardial Infarction: A Contemporary Community Perspective. Circ Heart Fail. 2016;9:e002460. [PMC free article] [PubMed] [Google Scholar]
- Chen J, Hsieh AF, Dharmarajan K, Masoudi FA, Krumholz HM. National trends in heart failure hospitalization after acute myocardial infarction for Medicare beneficiaries: 1998-2010. Circulation. 2013;128:2577–2584. [PMC free article] [PubMed] [Google Scholar]
- Gjesing A, Gislason GH, Køber L, Gustav Smith J, Christensen SB, Gustafsson F, Olsen AM, Torp-Pedersen C, Andersson C. Nationwide trends in development of heart failure and mortality after first-time myocardial infarction 1997-2010: A Danish cohort study. Eur J Intern Med. 2014;25:731–738. [PubMed] [Google Scholar]
- Hung J, Teng TH, Finn J, Knuiman M, Briffa T, Stewart S, Sanfilippo FM, Ridout S, Hobbs M. Trends from 1996 to 2007 in incidence and mortality outcomes of heart failure after acute myocardial infarction: a population-based study of 20,812 patients with first acute myocardial infarction in Western Australia. J Am Heart Assoc. 2013;2:e000172. [PMC free article] [PubMed] [Google Scholar]
- Desta L, Jernberg T, Löfman I, Hofman-Bang C, Hagerman I, Spaak J, Persson H. Incidence, temporal trends, and prognostic impact of heart failure complicating acute myocardial infarction. The SWEDEHEART Registry (Swedish Web-System for Enhancement and Development of Evidence-Based Care in Heart Disease Evaluated According to Recommended Therapies): a study of 199,851 patients admitted with index acute myocardial infarctions, 1996 to 2008. JACC Heart Fail. 2015;3:234–242. [PubMed] [Google Scholar]
- Guidry UC, Evans JC, Larson MG, Wilson PW, Murabito JM, Levy D. Temporal trends in event rates after Q-wave myocardial infarction: the Framingham Heart Study. Circulation. 1999;100:2054–2059. [PubMed] [Google Scholar]
- Velagaleti RS, Pencina MJ, Murabito JM, Wang TJ, Parikh NI, D’Agostino RB, Levy D, Kannel WB, Vasan RS. Long-term trends in the incidence of heart failure after myocardial infarction. Circulation. 2008;118:2057–2062. [PMC free article] [PubMed] [Google Scholar]
- Goldberg RJ, Spencer FA, Yarzebski J, Lessard D, Gore JM, Alpert JS, Dalen JE. A 25-year perspective into the changing landscape of patients hospitalized with acute myocardial infarction (the Worcester Heart Attack Study) Am J Cardiol. 2004;94:1373–1378. [PubMed] [Google Scholar]
- Ezekowitz JA, Kaul P, Bakal JA, Armstrong PW, Welsh RC, McAlister FA. Declining in-hospital mortality and increasing heart failure incidence in elderly patients with first myocardial infarction. J Am Coll Cardiol. 2009;53:13–20. [PubMed] [Google Scholar]
- Alabas OA, Allan V, McLenachan JM, Feltbower R, Gale CP. Age-dependent improvements in survival after hospitalisation with acute myocardial infarction: an analysis of the Myocardial Ischemia National Audit Project (MINAP) Age Ageing. 2014;43:779–785. [PubMed] [Google Scholar]
- Stone GW, Dixon SR, Grines CL, Cox DA, Webb JG, Brodie BR, Griffin JJ, Martin JL, Fahy M, Mehran R, et al. Predictors of infarct size after primary coronary angioplasty in acute myocardial infarction from pooled analysis from four contemporary trials. Am J Cardiol. 2007;100:1370–1375. [PubMed] [Google Scholar]
- Lewis EF, Moye LA, Rouleau JL, Sacks FM, Arnold JM, Warnica JW, Flaker GC, Braunwald E, Pfeffer MA. Predictors of late development of heart failure in stable survivors of myocardial infarction: the CARE study. J Am Coll Cardiol. 2003;42:1446–1453. [PubMed] [Google Scholar]
- Lewis EF, Velazquez EJ, Solomon SD, Hellkamp AS, McMurray JJ, Mathias J, Rouleau JL, Maggioni AP, Swedberg K, Kober L, et al. Predictors of the first heart failure hospitalization in patients who are stable survivors of myocardial infarction complicated by pulmonary congestion and/or left ventricular dysfunction: a VALIANT study. Eur Heart J. 2008;29:748–756. [PubMed] [Google Scholar]
- McAllister DA, Halbesma N, Carruthers K, Denvir M, Fox KA. GRACE score predicts heart failure admission following acute coronary syndrome. Eur Heart J Acute Cardiovasc Care. 2015;4:165–171. [PubMed] [Google Scholar]
- Sorajja P, Gersh BJ, Cox DA, McLaughlin MG, Zimetbaum P, Costantini C, Stuckey T, Tcheng JE, Mehran R, Lansky AJ, et al. Impact of multivessel disease on reperfusion success and clinical outcomes in patients undergoing primary percutaneous coronary intervention for acute myocardial infarction. Eur Heart J. 2007;28:1709–1716. [PubMed] [Google Scholar]
- Dudek D, Rakowski T, El Massri N, Sorysz D, Zalewski J, Legutko J, Dziewierz A, Rzeszutko L, Zmudka K, Piwowarska W, et al. Patency of infarct related artery after pharmacological reperfusion during transfer to primary percutaneous coronary intervention influences left ventricular function and one-year clinical outcome. Int J Cardiol. 2008;124:326–331. [PubMed] [Google Scholar]
- Mehta RH, Harjai KJ, Cox D, Stone GW, Brodie B, Boura J, O’Neill W, Grines CL. Clinical and angiographic correlates and outcomes of suboptimal coronary flow inpatients with acute myocardial infarction undergoing primary percutaneous coronary intervention. J Am Coll Cardiol. 2003;42:1739–1746. [PubMed] [Google Scholar]
- Costantini CO, Stone GW, Mehran R, Aymong E, Grines CL, Cox DA, Stuckey T, Turco M, Gersh BJ, Tcheng JE, et al. Frequency, correlates, and clinical implications of myocardial perfusion after primary angioplasty and stenting, with and without glycoprotein IIb/IIIa inhibition, in acute myocardial infarction. J Am Coll Cardiol. 2004;44:305–312. [PubMed] [Google Scholar]
- Wessler JD, Généreux P, Mehran R, Ayele GM, Brener SJ, McEntegart M, Ben-Yehuda O, Stone GW, Kirtane AJ. Which Intraprocedural Thrombotic Events Impact Clinical Outcomes After Percutaneous Coronary Intervention in Acute Coronary Syndromes?: A Pooled Analysis of the HORIZONS-AMI and ACUITY Trials. JACC Cardiovasc Interv. 2016;9:331–337. [PubMed] [Google Scholar]
- Planer D, Mehran R, Witzenbichler B, Guagliumi G, Peruga JZ, Brodie BR, Dudek D, Möckel M, Reyes SL, Stone GW. Prognostic utility of left ventricular end-diastolic pressure in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. Am J Cardiol. 2011;108:1068–1074. [PubMed] [Google Scholar]
- McGeoch R, Watkins S, Berry C, Steedman T, Davie A, Byrne J, Hillis S, Lindsay M, Robb S, Dargie H, et al. The index of microcirculatory resistance measured acutely predicts the extent and severity of myocardial infarction in patients with ST-segment elevation myocardial infarction. JACC Cardiovasc Interv. 2010;3:715–722. [PubMed] [Google Scholar]
- Fearon WF, Low AF, Yong AS, McGeoch R, Berry C, Shah MG, Ho MY, Kim HS, Loh JP, Oldroyd KG. Prognostic value of the Index of Microcirculatory Resistance measured after primary percutaneous coronary intervention. Circulation. 2013;127:2436–2441. [PMC free article] [PubMed] [Google Scholar]
- Johnson NP, Gould KL, Di Carli MF, Taqueti VR. Invasive FFR and Noninvasive CFR in the Evaluation of Ischemia: What Is the Future? J Am Coll Cardiol. 2016;67:2772–2788. [PubMed] [Google Scholar]
- Teunissen PF, de Waard GA, Hollander MR, Robbers LF, Danad I, Biesbroek PS, Amier RP, Echavarría-Pinto M, Quirós A, Broyd C, et al. Doppler-derived intracoronary physiology indices predict the occurrence of microvascular injury and microvascular perfusion deficits after angiographically successful primary percutaneous coronary intervention. Circ Cardiovasc Interv. 2015;8:e001786. [PubMed] [Google Scholar]
- Cuculi F, Dall’Armellina E, Manlhiot C, De Caterina AR, Colyer S, Ferreira V, Morovat A, Prendergast BD, Forfar JC, Alp NJ, et al. Early change in invasive measures of microvascular function can predict myocardial recovery following PCI for ST-elevation myocardial infarction. Eur Heart J. 2014;35:1971–1980. [PubMed] [Google Scholar]
- Patel N, Petraco R, Dall’Armellina E, Kassimis G, De Maria GL, Dawkins S, Lee R, Prendergast BD, Choudhury RP, Forfar JC, et al. Zero-Flow Pressure Measured Immediately After Primary Percutaneous Coronary Intervention for ST-Segment Elevation Myocardial Infarction Provides the Best Invasive Index for Predicting the Extent of Myocardial Infarction at 6 Months: An OxAMI Study (Oxford Acute Myocardial Infarction) JACC Cardiovasc Interv. 2015;8:1410–1421. [PubMed] [Google Scholar]
- Mollema SA, Nucifora G, Bax JJ. Prognostic value of echocardiography after acute myocardial infarction. Heart. 2009;95:1732–1745. [PubMed] [Google Scholar]
- Hung CL, Verma A, Uno H, Shin SH, Bourgoun M, Hassanein AH, McMurray JJ, Velazquez EJ, Kober L, Pfeffer MA, et al. Longitudinal and circumferential strain rate, left ventricular remodeling, and prognosis after myocardial infarction. J Am Coll Cardiol. 2010;56:1812–1822. [PubMed] [Google Scholar]
- Eitel I, Desch S, Fuernau G, Hildebrand L, Gutberlet M, Schuler G, Thiele H. Prognostic significance and determinants of myocardial salvage assessed by cardiovascular magnetic resonance in acute reperfused myocardial infarction. J Am Coll Cardiol. 2010;55:2470–2479. [PubMed] [Google Scholar]
- Larose E, Rodés-Cabau J, Pibarot P, Rinfret S, Proulx G, Nguyen CM, Déry JP, Gleeton O, Roy L, Noël B, et al. Predicting late myocardial recovery and outcomes in the early hours of ST-segment elevation myocardial infarction traditional measures compared with microvascular obstruction, salvaged myocardium, and necrosis characteristics by cardiovascular magnetic resonance. J Am Coll Cardiol. 2010;55:2459–2469. [PubMed] [Google Scholar]
- Eitel I, de Waha S, Wöhrle J, Fuernau G, Lurz P, Pauschinger M, Desch S, Schuler G, Thiele H. Comprehensive prognosis assessment by CMR imaging after ST-segment elevation myocardial infarction. J Am Coll Cardiol. 2014;64:1217–1226. [PubMed] [Google Scholar]
- Regenfus M, Schlundt C, Krähner R, Schönegger C, Adler W, Ludwig J, Daniel WG, Schmid M. Six-Year Prognostic Value of Microvascular Obstruction After Reperfused ST-Elevation Myocardial Infarction as Assessed by Contrast-Enhanced Cardiovascular Magnetic Resonance. Am J Cardiol. 2015;116:1022–1027. [PubMed] [Google Scholar]
- de Waha S, Desch S, Eitel I, Fuernau G, Zachrau J, Leuschner A, Gutberlet M, Schuler G, Thiele H. Impact of early vs. late microvascular obstruction assessed by magnetic resonance imaging on long-term outcome after ST-elevation myocardial infarction: a comparison with traditional prognostic markers. Eur Heart J. 2010;31:2660–2668. [PubMed] [Google Scholar]
- Mather AN, Fairbairn TA, Ball SG, Greenwood JP, Plein S. Reperfusion haemorrhage as determined by cardiovascular MRI is a predictor of adverse left ventricular remodelling and markers of late arrhythmic risk. Heart. 2011;97:453–459. [PubMed] [Google Scholar]
- Eitel I, Kubusch K, Strohm O, Desch S, Mikami Y, de Waha S, Gutberlet M, Schuler G, Friedrich MG, Thiele H. Prognostic value and determinants of a hypointense infarct core in T2-weighted cardiac magnetic resonance in acute reperfused ST-elevation-myocardial infarction. Circ Cardiovasc Imaging. 2011;4:354–362. [PubMed] [Google Scholar]
- Carrick D, Haig C, Rauhalammi S, Ahmed N, Mordi I, McEntegart M, Petrie MC, Eteiba H, Hood S, Watkins S, et al. Prognostic significance of infarct core pathology revealed by quantitative non-contrast in comparison with contrast cardiac magnetic resonance imaging in reperfused ST-elevation myocardial infarction survivors. Eur Heart J. 2016;37:1044–1059. [PMC free article] [PubMed] [Google Scholar]
- Wollenweber T, Roentgen P, Schäfer A, Schatka I, Zwadlo C, Brunkhorst T, Berding G, Bauersachs J, Bengel FM. Characterizing the inflammatory tissue response to acute myocardial infarction by clinical multimodality noninvasive imaging. Circ Cardiovasc Imaging. 2014;7:811–818. [PubMed] [Google Scholar]
- Rischpler C, Dirschinger RJ, Nekolla SG, Kossmann H, Nicolosi S, Hanus F, van Marwick S, Kunze KP, Meinicke A, Götze K, et al. Prospective Evaluation of 18F-Fluorodeoxyglucose Uptake in Postischemic Myocardium by Simultaneous Positron Emission Tomography/Magnetic Resonance Imaging as a Prognostic Marker of Functional Outcome. Circ Cardiovasc Imaging. 2016;9:e004316. [PMC free article] [PubMed] [Google Scholar]
- Lapa C, Reiter T, Werner RA, Ertl G, Wester HJ, Buck AK, Bauer WR, Herrmann K. [(68)Ga]Pentixafor-PET/CT for Imaging of Chemokine Receptor 4 Expression After Myocardial Infarction. JACC Cardiovasc Imaging. 2015;8:1466–1468. [PubMed] [Google Scholar]
- Stubbs P, Collinson P, Moseley D, Greenwood T, Noble M. Prognostic significance of admission troponin T concentrations in patients with myocardial infarction. Circulation. 1996;94:1291–1297. [PubMed] [Google Scholar]
- Reinstadler SJ, Feistritzer HJ, Reindl M, Klug G, Mayr A, Mair J, Jaschke W, Metzler B. Combined biomarker testing for the prediction of left ventricular remodelling in ST-elevation myocardial infarction. Open Heart. 2016;3:e000485. [PMC free article] [PubMed] [Google Scholar]
- Azab B, Zaher M, Weiserbs KF, Torbey E, Lacossiere K, Gaddam S, Gobunsuy R, Jadonath S, Baldari D, McCord D, et al. Usefulness of neutrophil to lymphocyte ratio in predicting short- and long-term mortality after non-ST-elevation myocardial infarction. Am J Cardiol. 2010;106:470–476. [PubMed] [Google Scholar]
- Núñez J, Núñez E, Bodí V, Sanchis J, Miñana G, Mainar L, Santas E, Merlos P, Rumiz E, Darmofal H, et al. Usefulness of the neutrophil to lymphocyte ratio in predicting long-term mortality in ST segment elevation myocardial infarction. Am J Cardiol. 2008;101:747–752. [PubMed] [Google Scholar]
- Seropian IM, Sonnino C, Van Tassell BW, Biasucci LM, Abbate A. Inflammatory markers in ST-elevation acute myocardial infarction. Eur Heart J Acute Cardiovasc Care. 2016;5:382–395. [PubMed] [Google Scholar]
- Sato A, Aonuma K, Imanaka-Yoshida K, Yoshida T, Isobe M, Kawase D, Kinoshita N, Yazaki Y, Hiroe M. Serum tenascin-C might be a novel predictor of left ventricular remodeling and prognosis after acute myocardial infarction. J Am Coll Cardiol. 2006;47:2319–2325. [PubMed] [Google Scholar]
- Sato A, Hiroe M, Akiyama D, Hikita H, Nozato T, Hoshi T, Kimura T, Wang Z, Sakai S, Imanaka-Yoshida K, et al. Prognostic value of serum tenascin-C levels on long-term outcome after acute myocardial infarction. J Card Fail. 2012;18:480–486. [PubMed] [Google Scholar]
- Voors AA, von Haehling S, Anker SD, Hillege HL, Struck J, Hartmann O, Bergmann A, Squire I, van Veldhuisen DJ, Dickstein K. C-terminal provasopressin (copeptin) is a strong prognostic marker in patients with heart failure after an acute myocardial infarction: results from the OPTIMAAL study. Eur Heart J. 2009;30:1187–1194. [PubMed] [Google Scholar]
- Khan SQ, Dhillon OS, O’Brien RJ, Struck J, Quinn PA, Morgenthaler NG, Squire IB, Davies JE, Bergmann A, Ng LL. C-terminal provasopressin (copeptin) as a novel and prognostic marker in acute myocardial infarction: Leicester Acute Myocardial Infarction Peptide (LAMP) study. Circulation. 2007;115:2103–2110. [PubMed] [Google Scholar]
- Weir RA, Petrie CJ, Murphy CA, Clements S, Steedman T, Miller AM, McInnes IB, Squire IB, Ng LL, Dargie HJ, et al. Galectin-3 and cardiac function in survivors of acute myocardial infarction. Circ Heart Fail. 2013;6:492–498. [PubMed] [Google Scholar]
Articles from World Journal of Cardiology are provided here courtesy of Baishideng Publishing Group Inc
Author information
Thomas J Cahill, Rajesh K Kharbanda, Oxford Heart Centre, John Radcliffe Hospital, Oxford OX3 9DU, United Kingdom
Author contributions: Cahill TJ and Kharbanda RK both devised, drafted and revised the manuscript.
Correspondence to: Rajesh K Kharbanda, Professor, Oxford Heart Centre, John Radcliffe Hospital, Headley Way, Oxford OX3 9DU, United Kingdom. Email: rajesh.kharbanda@ouh.nhs.uk
Telephone: +44-1865-220325 Fax: +44-1865-740409
Received 2016 Oct 30; Revised 2017 Jan 20; Accepted 2017 Mar 12.
Copyright and License information
Copyright ©The Author(s) 2017. Published by Baishideng Publishing Group Inc. All rights reserved.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/