Disseminated Superficial Porokeratosis with Dermal Amyloid Deposits
M. Carlesimo
aU.O.C. Dermatology, II Unit University of Rome ‘Sapienza’, Rome, Italy
A. Rossi
bDepartment of Dermatology and Venereology, I Unit University of Rome ‘Sapienza’, Rome, Italy
L. Fidanza
aU.O.C. Dermatology, II Unit University of Rome ‘Sapienza’, Rome, Italy
A. Narcisi
aU.O.C. Dermatology, II Unit University of Rome ‘Sapienza’, Rome, Italy
M. La Pietra
aU.O.C. Dermatology, II Unit University of Rome ‘Sapienza’, Rome, Italy
E. Mari
aU.O.C. Dermatology, II Unit University of Rome ‘Sapienza’, Rome, Italy
C. Cacchi
cDepartment of Histopathology, II Unit University of Rome ‘Sapienza’, Rome, Italy
G. Camplone
aU.O.C. Dermatology, II Unit University of Rome ‘Sapienza’, Rome, Italy
Abstract
Only 6 cases with an association of disseminated superficial porokeratosis with dermal amyloid deposits are reported in the literature. We present the case of a 76-year-old woman who presented with a disseminated superficial porokeratosis. Histological examination revealed amyloid deposits in the upper dermis, which were typed with routine HE stains, Congo red stains and anticytokeratin antibodies (AE1-AE3 and CK5). Positive staining with Congo red and, moreover, with CK5 (a cytokeratin strongly represented in the basal cell layer of the epidermis) indicates an epidermal origin of this protein.
Introduction
Porokeratoses are a group of cutaneous entities of unknown etiology, characterized by disordered epidermal keratinization and, at histological examination, by the presence of coronoid lamella, a column of parakeratotic cells extending through the stratum corneum [1].
Disseminated superficial porokeratosis (DSP) is a distinct form of porokeratosis that, clinically, is characterized by numerous, small, superficial keratotic papules with verrucoid aspects, an atrophic center and peripheral keratotic border. Sometimes DSP can be associated with dermal amyloid deposits [1]. Only 7 cases of this association have been described in the literature (table 1) [2].
Table 1
DSP with amyloid deposits: a summary of the reported cases
| Piamphongstant and Sittapairoachana, 1974 [5] | Stefanato etal., 1993 [6] | Yasuda etal., 1996 [7] 1st case | Yasuda etal., 1996 [7] 2nd case | Aman tea etal., 1998 [3] | Demitsu and Okada, 1999 [8] | Kim et al., 2000 [2] | |
|---|---|---|---|---|---|---|---|
| Age/sex | 51/F | 32/F | 63/M | 60/M | 72/M | 63/M | 88/M |
| Age at onset | 6th decade | 4th decade | 5th decade | 6th decade | 7th decade | adolescence | 7th decade |
| Duration | 7 years | 1 month | 20 years | 2 years | 3-4 years | n.d. | 20 years |
| Pruritus | n.d. | none | slight | present | none | n.d. | severe |
| Family history | n.d. | negative | n.d. | n.d. | negative | negative | daughter |
| Treatment | n.d. | topical steroid/etretinate | topical DMSO | topical DSMO topical steroid | n.d. | cryosurgery | topical steroid antihistamine cryosurgery |
| Results | not improved | improved | worsened | improved | improved |
n.d. = Not described; DSMO = dimethyl sulfoxide.
We report a eighth case of this type and an atypical immunohistochemical method used to characterize the amyloid substance.
Case Report
A 76-year-old woman was referred to our Department in June 1999. She reported a 20 years’ history of an erythematous and squamo-papular eruption located on the extremities, without itching or pain. The patient had noted a progressive extension of these lesions in the last year and an exacerbation of the lesions during the summer. Upon physical examination, numerous papules with a size of less than 15 mm, an atrophic center and hyperkeratotic borders were distributed on the neck, chest and extremities. Palms, soles, mucous membranes and nails were spared, and the patient denied any family history of similar skin lesions. She had no other symptoms and laboratory tests were all normal or negative.
We examined a skin biopsy that revealed atrophy of the epidermis with hyperorthokeratosis and presence of homogeneous and compact cornoid lamella of keratin, typically oriented sideways with respect to the epidermal tissue (fig. 1 a). The superficial dermis showed a slight chronic inflammatory infiltrate with vascular ectasia and edema. Moreover, in the papillary dermis a compact deposit of a cellular eosinophilic material suspicious for amyloid was observed (fig. 1b).
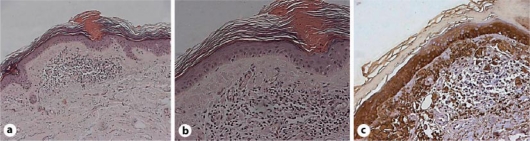
a Hyperkeratosis and coronoid lamella. b Eosinophilic, acellular deposits in the dermis. c Strong immunoreaction of amyloid for CK5.
To demonstrate the epithelial origin of the amyloid, we performed two immunohistochemical stainings by using specific MoAbs to pan-cytokeratin AE1-AE3. Interestingly, there was a striking difference between the two results: in fact, the acellular deposits, mentioned above, were negative for CKAE1-AE3, whereas a strong immunoreactivity was demonstrated for CK5, supporting a specific origin of the amyloid substance from the basal cell layer of the epidermis (fig. 1c).
Discussion
In 1937, Andrews first described DSP and introduced this term to indicate a clinical variant of Mibelli’s porokeratosis. Later, Chernoski and Freeman proposed a possible actinic etiology of this dermatosis and coined the term disseminated superficial ‘actinic’ porokeratosis (DSAP). Nowadays, this term is generally accepted in European dermatological literature, and this definition is based on clinical and histological findings. Dermatological manifestations are typically confined to sun exposed areas, with actinic induction and exacerbations [2].
In the literature, two types of localized cutaneous amyloidosis (LCA) are described: primary LCA (macular amyloidosis and lichen amyloidosis), which is not associated with other dermatoses or systemic involvement, and secondary LCA, which is associated with inflammatory, hamartomatous or neoplastic skin disorders [2].
The mechanism by which DSP induces dermal amyloid deposits is not clear, but Piamphongstant et al. first suggested that this process can derive from degenerated epidermal keratinocytes [2]. We hypothesize that a mutant keratinocyte clone is responsible for induction of porokeratotic lesions, because these necrotic epidermal keratinocytes (colloid bodies) might be transformed into amyloid by dermal macrophages and fibroblasts [3]. Immunohistochemical staining has shown an overexpression of p53 protein in porokeratotic lesions; this is a tumor suppressor protein, an important gatekeeper and effector of the cell cycle. Mutations of the p53 gene in all forms of porokeratosis, also in DSAP, create a permissive state of uncoordinated cell cycling, and predispose cells to death [4].
In our case, the lack of systemic involvement led to our hypothesis of a secondary dermal deposition of amyloid proteins and the existence of a close relationship between these two processes. This hypothesis was confirmed by positive staining with Congo red and immunohistochemical staining with the anticytokeratin MoAbs strongly positive for CK5, just below the epidermal porokeratotic zone in close proximity to the cornoid lamella. This cytokeratin is, in fact, strongly represented in the basal cell layer and these results indicate that the dermal deposits were amyloid originating from the epidermis (type II keratin).
Nowadays, more studies are necessary to clarify the exact mechanism that leads to secondary deposition of dermal amyloid in porokeratotic disease and the frequency of this association. We also suggest utilization of MoAbs for CK5 as a first-line target in these conditions, encouraged by the immediate and strong positivity of this protein in immunohistochemical studies, whereas pan-cytokeratin at first produced negative results.
References
Content retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2895207/.